



Why is LSD so potent and why does it last so long?
The famous 5HT2A receptor “Lid” paper…


The discovery of the psychedelic properties of LSD by Albert Hofmann represented a remarkable event from both a cultural and pharmacological perspective. As well as seeding the psychedelic revolution that would bloom in the decades that followed, LSD itself is something of a chemical and pharmacological marvel. As well as being one of the most potent psychedelic molecules known, with an active psychedelic dose as low as 50 micrograms (millionths of a gram), it also boasts one of the longest effect durations — up to around 15 hours.
Both the potency and effect duration of LSD remained somewhat mysterious since its discovery in 1943. How could such a tiny quantity of drug, with so few molecules actually reaching the brain, have such powerful and long-lasting effects on consciousness?
In the last post, I discussed a brand new landmark paper that revealed how the classic psychedelics induce neuroplasticity by activating intracellular 5HT2A receptors:

In this post I want to discuss another landmark paper — this time from 2017 — that finally provided a satisfying explanation for LSD’s hitherto seemingly miraculous abilities.

Here’s the paper you can download for free:
https://www.cell.com/fulltext/S0092-8674(16)31749-4
First, some neuropharmacology background:
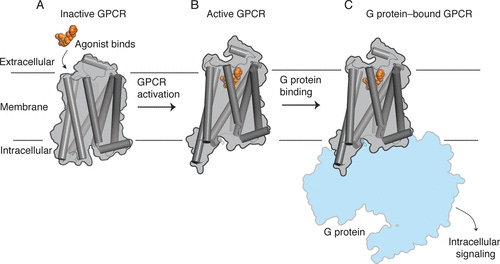
After oral ingestion, LSD is rapidly absorbed into the blood and brain, reaching a peak effect 2-3 hours later, before slowly declining over the next 8-12 hours. Like all classic psychedelics, LSD elicits its effects primarily by binding and activating the serotonin 5HT2A receptor heavily expressed in neurons in the deeper layers of the cortex. Binding of LSD to the receptor activates signalling pathways inside the neuron responsible for the changes in the neuron’s behaviour (and ultimately the psychedelic effects).
Receptors are flexible proteins that shift between conformational states. In the simplest case, a receptor will have an inactive conformation that doesn’t interact with intracellular signalling proteins and is thus “silent” and an active conformation that’s able to bind to these signalling proteins and activate the pathways that elicit the effects on the neuron’s function. The 5HT2A receptor, for example, interacts with a signalling protein called Gq (from a family of signalling proteins known as the G-proteins — hence the 5HT2A receptor is known as a G-protein coupled receptor, GPCR), which ultimately leads to depolarisation of the neuron (makes it more excitable and likely to fire). This is partly what engenders the psychedelic effect at the cortical level.
However, the 5HT2A receptor can also interact with a different signalling pathway — the beta-arrestin pathway — which has also been shown to be important in generating psychedelic effects (LINK). Although the precise mechanism of this remains unclear, the arrestins are known to be important structural scaffolds for assemblies of signalling proteins, so they might have a role in organising signalling through the neuron’s interior.

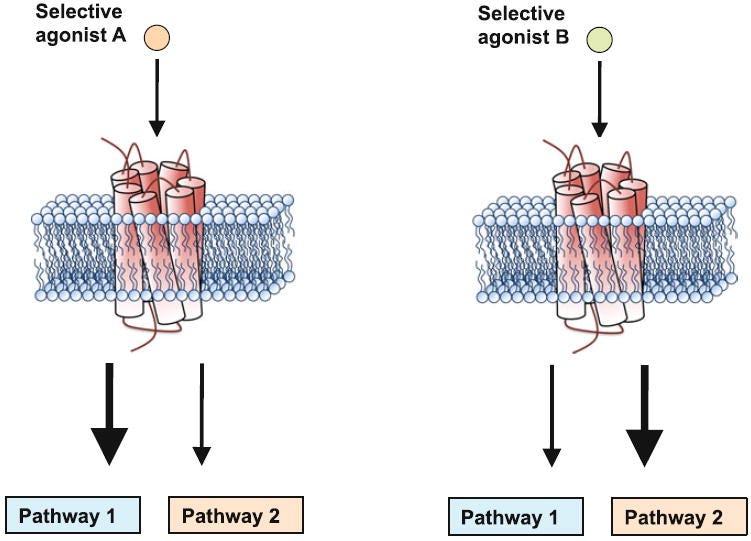
The particular pattern of Gq and beta-arrestin signalling will depend upon the active conformation that the receptor adopts, which can be controlled by the binding of different molecules. Crucially, when LSD binds to the 5HT2A receptor, it activates a particular balance of both Gq and beta-arrestin signalling. Different molecules, depending on their molecular structure, will elicit different patterns of signalling that may or may not induce psychedelic effects — this is known as signalling bias (or functional selectivity). Serotonin, which is not psychedelic, is known as a balanced agonist, since it doesn’t favour activation of the Gq pathway over the beta-arrestin pathway, whereas the classic psychedelics are biased agonists, since they favour one pathway over the other (by shifting the receptor into distinct conformations when they bind).
This figure shows the “arrestin bias” (how much the molecule activates the beta-arrestin pathway over the Gq pathway) for a number of molecules, including LSD, at the 5HT2B receptor (related to the 5HT2A receptor).

Molecules don’t bind to a receptor and then simply sit there permanently, except in the rare case that the molecule can form a strong covalent bond with the receptor (the lethal nerve agent VX, for example, forms a covalent bond in the binding site of the enzyme acetylcholinesterase). They bind and unbind to the receptor at different rates, known as the ‘on’ (k1) and ‘off’ (k2) rates, depending on their interactions with the receptor. This can be formalised as an equilibrium:
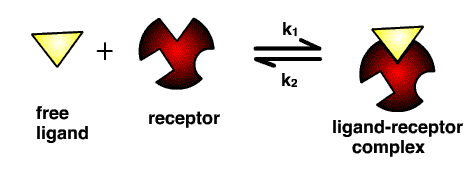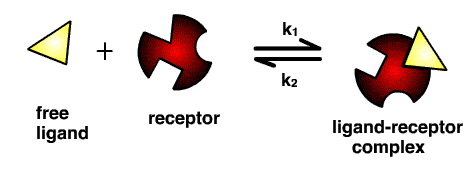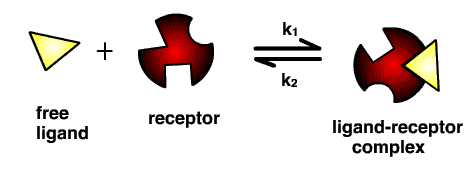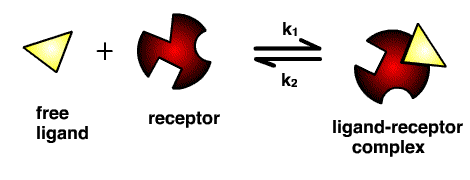
The residence time (also known as the dwell time) is the reciprocal of the ‘off’ rate, k2 (i.e. 1/k2) and represents the time a molecule spends bound to the receptor before dissociating (measured in seconds or minutes). This is an important measure, since molecules with the same ‘on’ rate and rate of clearance from the blood can have very different effect durations depending on their residence times in the receptor.
In this landmark paper, Bryan Roth’s group at the University of North Carolina School of Medicine were able to obtain a crystal structure (the gold standard of protein structural imaging) of the 5HT2B receptor (closely related to the 5HT2A receptor) bound to a molecule of LSD. This allowed them to directly visualise the interactions between the LSD molecule and the receptor, as well as the conformation adopted by the receptor in the bound state. They also used computational molecular modelling to predict how this structure translates to the 5HT2A receptor. Broadly, the team discovered two unique features of LSD’s interaction with the 5HT2A/B receptor that might account for both its extreme potency (the dose required to elicit a psychedelic effect) and duration of action:
The diethyl amide side chain adopts a unique position and orientation in the receptor binding site that is optimal for receptor signalling.
LSD has an extremely long residence time, owing to a portion of the receptor (the authors refer to this as a ‘lid’) that folds over the LSD molecule and is locked shut by a unique latch mechanism.
When LSD binds to the 5HT2A receptor (or any molecule to any receptor for that matter), areas of the molecule or particular chemical groups (such as amine or hydroxyl groups) interact with amino acids within the receptor protein chain. We can think of these amino acids as “microswitches” inside the ligand binding site of the receptor: the conformation the receptor adopts will depend on which of these microswitches the molecule binds and pushes/pulls on as it drops into the receptor. Many of these microswitches within the 5HT2A receptor have been identified, but I’ll discuss only a few known to be particularly important for the binding of LSD (and other classic psychedelics).

Tryptamines, such as DMT and serotonin, contain an indole ring with a 2-carbon chain that terminates in an amine (the aminoethyl side chain). LSD is essentially rigidified tryptamine — whereas the tryptamine side chain swings loose in other psychedelic tryptamines, in LSD (and related molecules) it’s locked in position by an additional two rings that form the ergoline ring system.

Flexible molecules are better able to rotate their bonds to optimise their interactions in a ligand binding site — they can “relax” into position. However, at the same time, the extra degrees of rotational freedom mean they have a lot of different orientations to search through to find the optimal one that minimises their energy. Furthermore, once they’ve found their position in the binding site, rotation of a single bond (perhaps caused by the natural movement of the receptor) can cause them to slip out again. Flexibility is a double-edged sword. Rigid molecules, in contrast, don’t have that opportunity, since their shape is fixed. As such, they must possess the correct shape to fit into the binding site from the outset, otherwise the site will simply reject them. However, if the molecule is rigidified in an optimal orientation, the molecule will slip effortlessly into the binding site and rapidly form the interactions required for receptor activation.
Remarkably, LSD seems to have found the optimal way to rigidify the tryptamine side chain to achieve an almost perfect match with the crucial amino acids in the ligand binding site: As the indole ring slides into position, guided by the flat electron-rich rings of phenylalanine (Phe) and tryptophan (Trp) within the receptor binding site, the indole nitrogen forms a hydrogen bound with a well-placed serine (Ser), and the side chain amine becomes perfectly positioned to interact with a critical aspartate (Asp).
However, what’s particularly interesting about LSD is the pair of ethyl groups (-CH2CH3) on the amide that give LSD its name (lysergic acid diethyl amide). Compared to LSA (lysergic acid amide), which has a free amide (no groups attached to the amide nitrogen), LSD is around ten times more potent — LSD is active above around 50 micrograms, compared to around 500 micrograms for LSA. Being the only structural difference between the two molecules, clearly these two ethyl groups have an important role in the dramatic difference in potency: Any changes to this group — such as increasing the length of the carbon chains or altering their structure in other ways — also cause a dramatic loss in potency. LSD is just right.
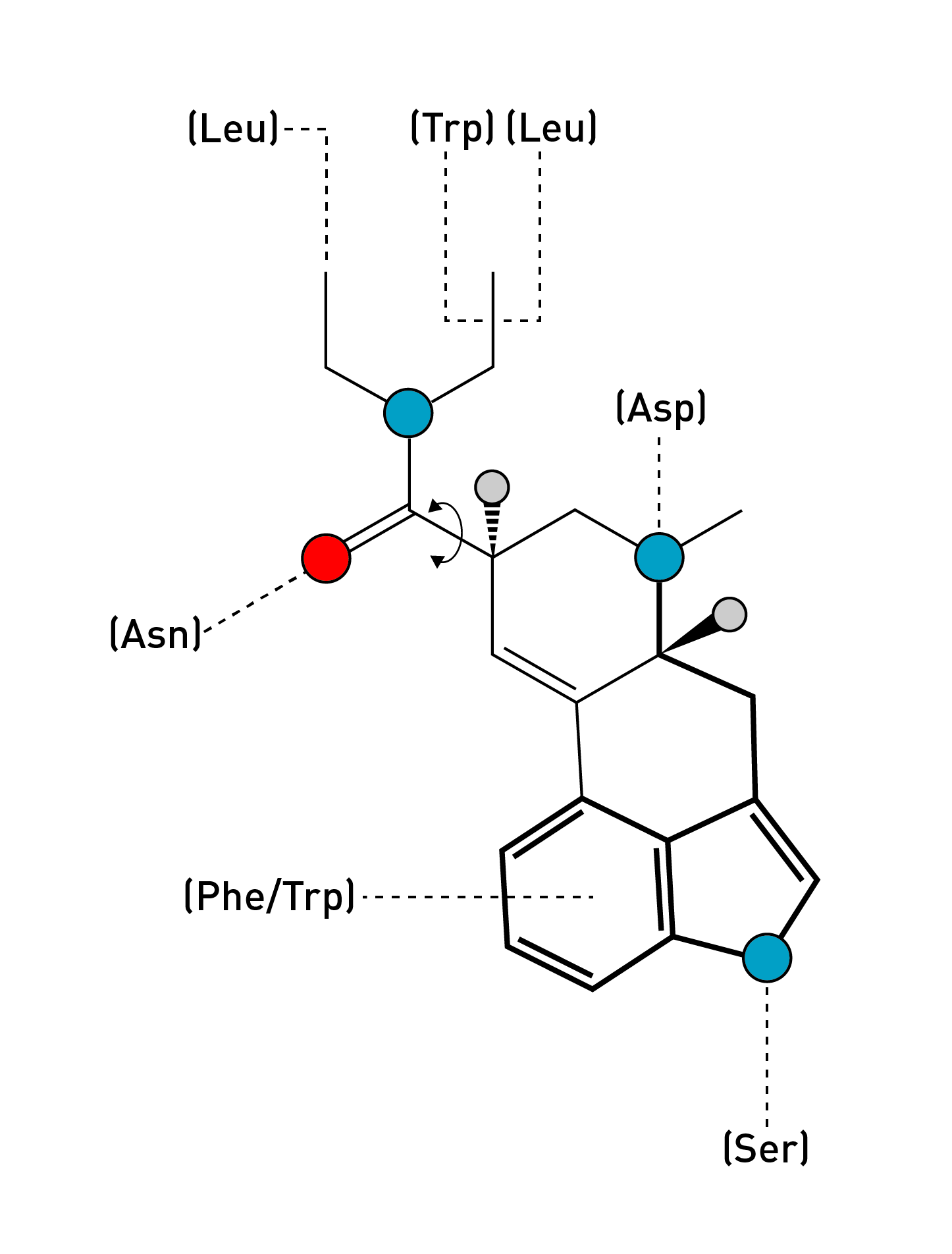
The ethyl groups themselves always tend to point away from each other (to minimise clashing interactions). However, the bond connecting the diethylamide group to the ergoline system can rotate, altering their spatial relationship to each other and thus the overall shape of the molecule. This creates two distinct conformations that the LSD molecule tends to adopt:
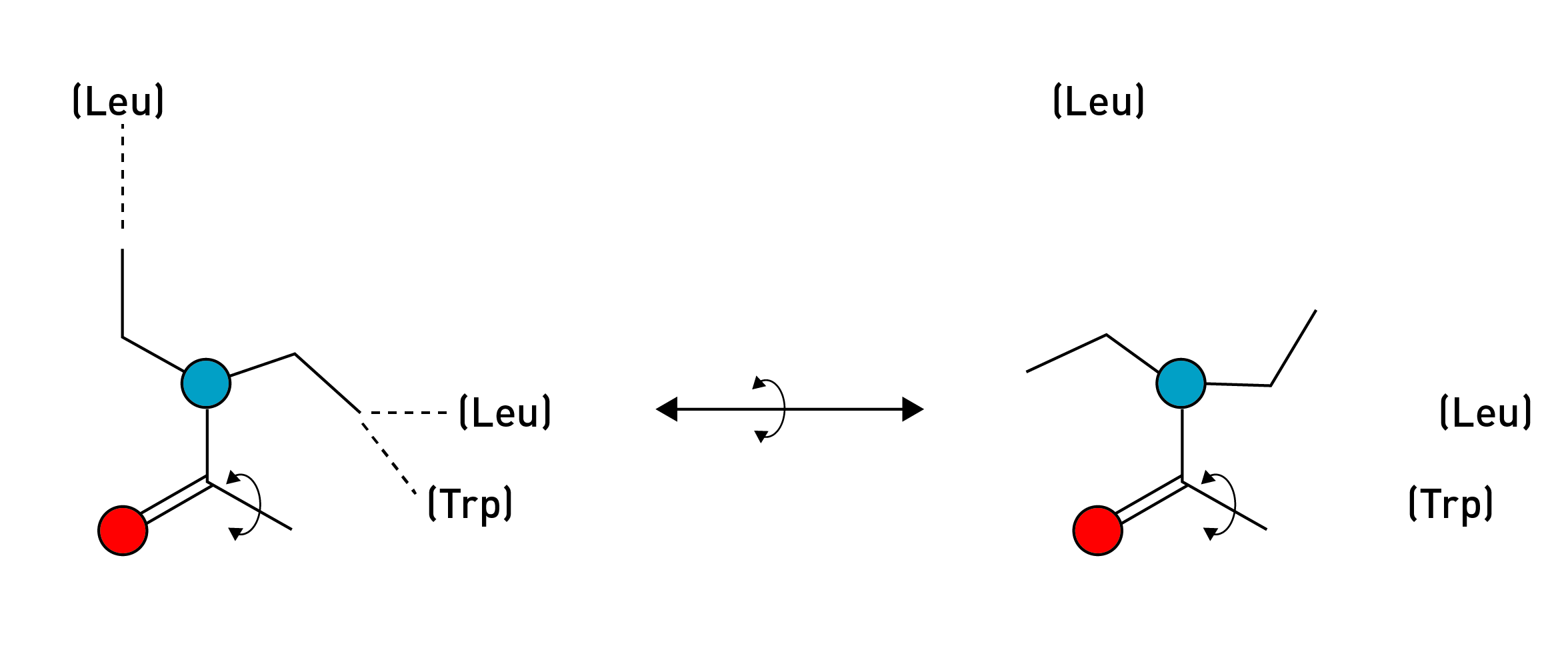
This is easier to visualise than to explain: In the diagram below from the paper, the two conformations of LSD are overlaid in yellow and pink. Notice how the ethyl groups in the yellow molecule point away from each other and in the opposite directions to those of the pink molecule. These are the two rotational conformations of LSD.

From the crystal structure of LSD bound to the 5HT2B receptor, the authors noticed that LSD adopted the ‘pink’ conformation rather than the ‘yellow’. They thus surmised that this conformation was important in stabilising the LSD molecule in the binding site and activating the receptor. Indeed, in the ‘pink’ conformation, one ethyl group becomes perfectly positioned to form an interaction with a leucine amino acid, and the other to interact with a tryptophan and another leucine. Rotating into the yellow conformation would disrupt these interactions. To test this, they created two molecules that “locked” the ethyl groups in either the ‘yellow’ or ‘pink’ conformation by making them part of a rigid 4-membered ring that can’t flip between states.

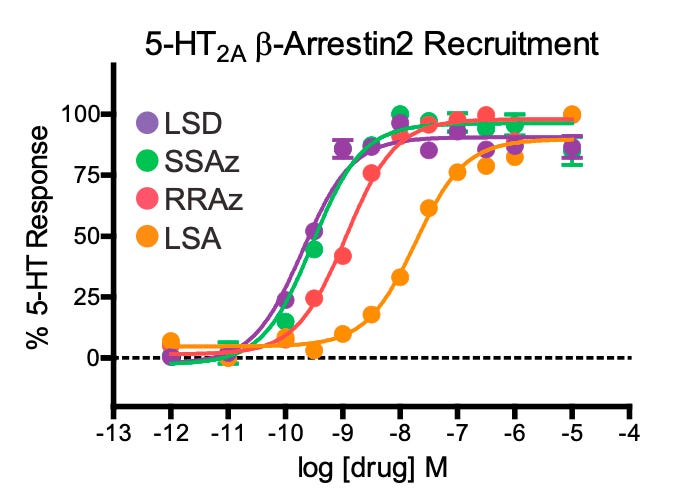
(S,S)-Azetidide (SSAz) most closely resembles the ‘pink’ conformation of LSD, whereas (R,R)-Azetidide (RRAz) resembles the ‘yellow’ conformation. Since LSD adopts the ‘pink’ conformation in the receptor binding site, they predicted that SSAz ought to be a more potent activator of the 5HT2A receptor than RRAz (which would be unable to form the required interactions in the binding site). Interestingly, they found that both SSAz and RRAz were almost equally potent in activating the Gq signalling pathway, whilst RRAz was much weaker in activating the beta-arrestin pathway. This suggests the ‘pink’ conformation of the diethyl groups is important in eliciting the particular pattern of signalling that engenders the psychedelic effect. LSA, which doesn’t contain the ethyl groups at all, was even less potent at activating beta-arrestin signalling. Overall, this demonstrates the importance of these two ethyl groups in the unusual potency of the LSD molecule.
Next, the authors measured the rates at which LSD bound and unbound to the 5HT2A receptor and found that the off-rate was extremely small (0.005 per min), meaning LSD has a residence time of more than 3.5 hours!
What could be causing LSD to remain stuck in the receptor binding site for so long?
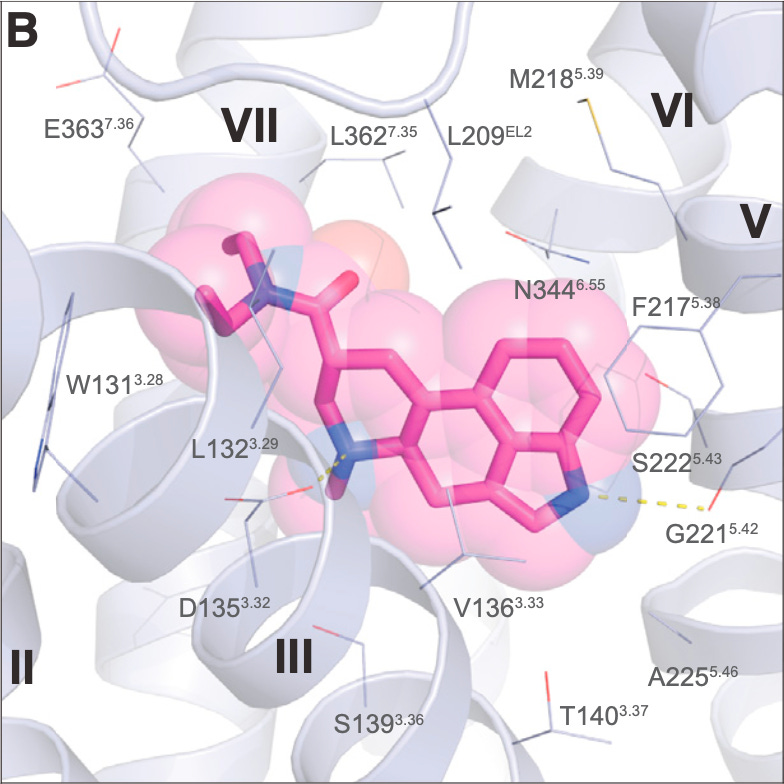
In the LSD-receptor crystal structure, the authors noticed that a portion of the receptor protein chain folded over the LSD molecule to form a “lid” that might hinder the molecule’s ability to leave the receptor once bound. They also noticed that a particular leucine amino acid on the underside of the lid formed a number of interactions with the LSD molecule, leading them to hypothesise that this leucine might act as a “latch” that keeps the lid shut, preventing LSD from escaping. In support of this hypothesis, molecular simulations of the receptor showed that this lid can flip rapidly between open and closed states, but does so much less frequently when LSD is bound.

To test this further, the authors mutated this “latch” leucine amino acid to an alanine (which is unable to interact with the LSD molecule) in both the simulated and actual receptor. In the simulated mutant receptor, the “lid” flipped between open and closed states much more frequently whether or not LSD was present, indicating that the molecule was unable to “close the latch” when the leucine wasn’t present. In the real mutant receptor, the residence time of LSD was reduced 10-fold, further supporting the hypothesis that this lid latch was important in trapping the molecule in the receptor binding site. Furthermore, LSD was much less potent in stimulating the beta-arrestin pathway in the mutant receptor, suggesting that the long residence time is important in maximising the intracellular signalling central to LSD’s psychedelic effects.
So, overall, this landmark study was able to explain both the extreme potency and long effect duration of LSD compared to most other psychedelics. Specifically, the diethyl groups unique to LSD adopt an orientation in the ligand binding site of the 5HT2A receptor that allows them to interact with specific leucine and tryptophan amino acids that shift the receptor into an optimal conformation to stimulate the particular pattern of Gq and beta-arrestin signalling that underpins the psychedelic effect. Furthermore, once in the receptor binding site, LSD interacts with a leucine on the receptor lid and effectively locks it shut from the inside! This allows it to remain in the binding site and actively signalling for several hours before eventually escaping.
Thank you for elucidating this paper. Greatly appreciated.
Do we currently have a clear understanding of why different signaling biases result in the various subjective effects of, for example, LSD versus LSA? Or do we only know that this is the case without further insight into how and why it happens?
This does not explain "long LSD" - like long COVID :)
As I rose up the ranks of the scientific and corporate world I started to wonder: where did all the acid heads go? Very few people who regularly dropped acid avoided dropping out.