



The story of Bromo-DragonFLY
The world’s most potent psychedelic…???

There are a number of ways to design a new drug molecule. Historically, the most common method was by brute force: take a molecule we know has some desired psychoactivity activity and manipulate its structure — adding, removing, or changing chemical groups or even its skeletal structure — and test each new molecule for activity. There is a whole field of chemistry, known as combinatorial chemistry, devoted to this: make as many variants of a molecule as possible (sometimes hundreds or more) and screen each one for biological activity. In the case of psychoactive drugs, in which their activity usually depends on binding and activating (or blocking) a particular receptor, this would often be by measuring the molecule’s affinity for the receptor (which can now be automated). Molecules that show promising binding properties can then be followed up with further testing, such as activity in an animal model and then, ultimately, in humans.
At the opposite end of the spectrum is a modern rational drug design approach. This has only become possible with modern x-ray crystallography techniques, as well as computational modelling of the interactions between receptors and drug molecules. As I discussed in the last post, obtaining a crystal of a molecule bound to a receptor is the Holy Grail for structural biologists. Whenever the crystal structure of a new receptor is obtained, you’ll invariably see it published in the highest echelon of academic journals. Generally that means either Science, Nature, or Cell.

Once we have a fairly well-established picture of the important interactions between a molecule and a receptor, it’s possible to design molecules with the requisite structure to bind and activate the receptor. Modern computational modelling software can also simulate the docking of a molecule to a receptor before it’s actually synthesised in the lab.
Bromo-DragonFLY is a beautiful example of drug design performed by one of the best in the business — Dave Nichols at Purdue — and shows how a deep understanding of drug-receptor interactions can be used to generate molecules of truly exceptional potency.
Important note: When we say potent, we mean the dose required to elicit the desired effect or the concentration of drug required to stimulate a particular response from a receptor. Potency does not refer to the intensity of the subjective effect. DMT, for example, is an extremely intense psychedelic, active in the tens of milligrams range, but it’s not particularly potent compared to, say, LSD, which is active at doses as low as 25 micrograms.
Peerless psychedelic chemist Alexander “Sasha” Shulgin’s approach to producing new drugs sat somewhere between brute force and modern rational design: Whilst he didn’t generally have access to the crystal structures and computational modelling software commonly used today, his knowledge and understanding of structure-activity relationships was encyclopaedic and, as such, he didn’t merely tinker with making new molecules but usually had a very good rationale for making a particular molecule based on other molecules he’d either created or worked with. Spend some time reading the “Extensions and Commentary” sections of each entry in PIHKAL and TIHKAL to really understand how the mind of a genius chemical pharmacologist works.
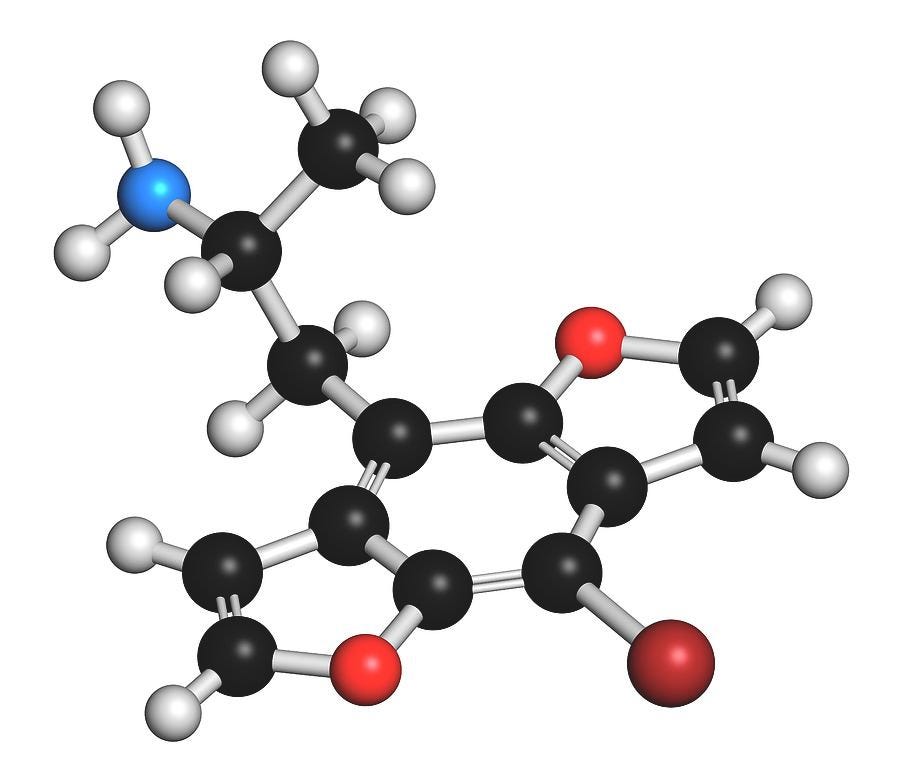
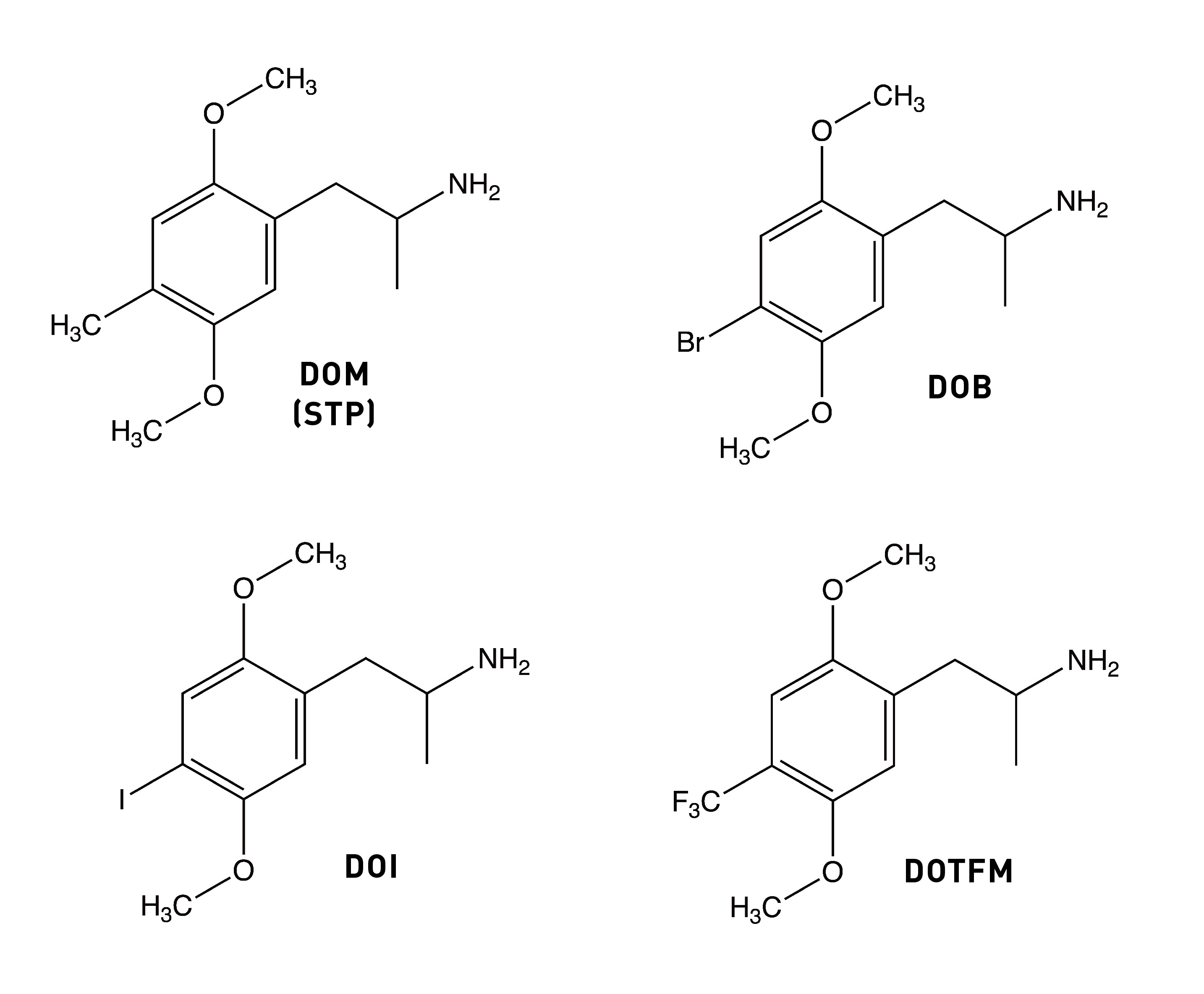
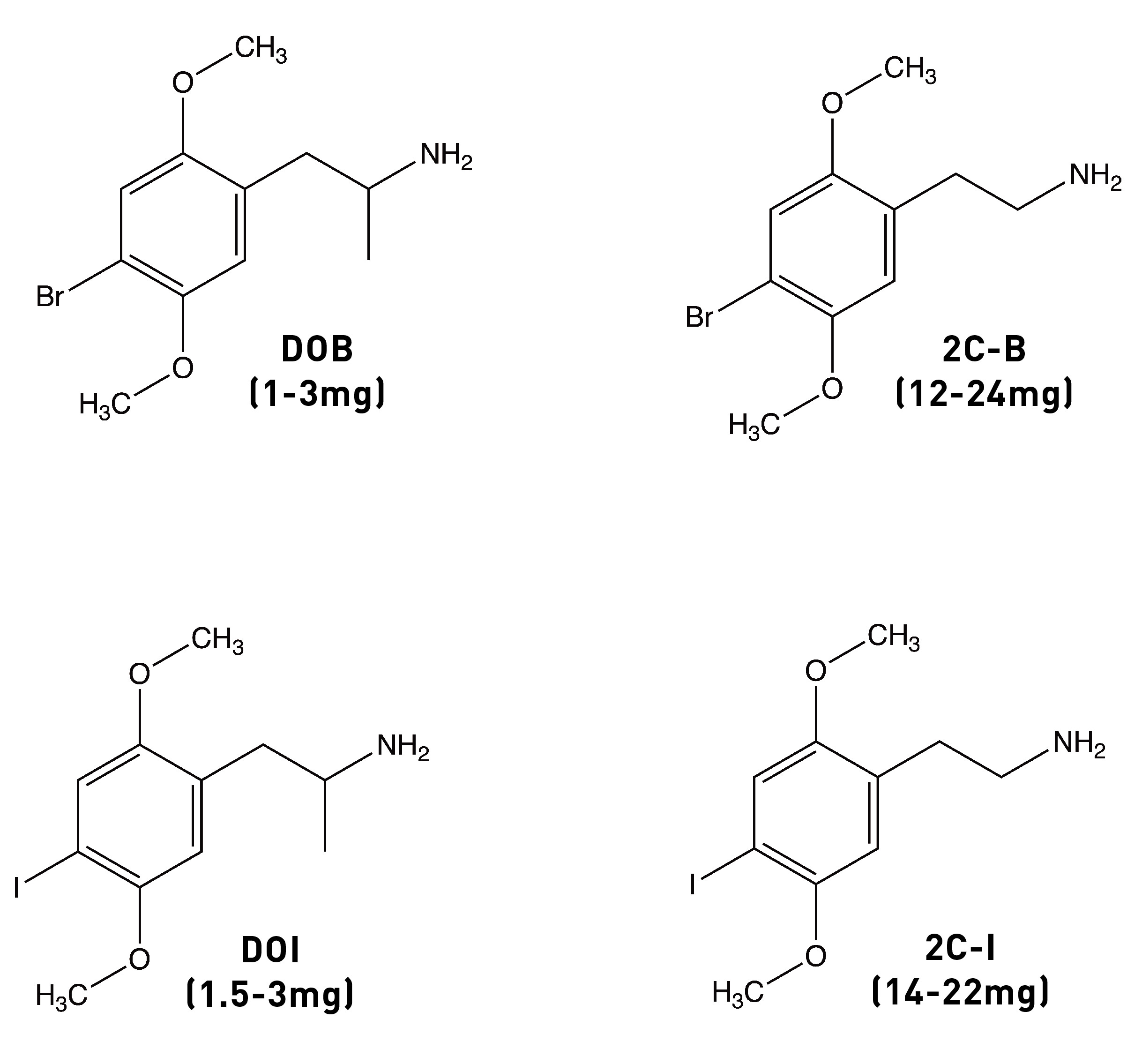
The story of Bromo-DragonFLY really starts in 1967, when a powerful psychedelic known as STP began circulating on the streets of the Haight-Ashbury in San Francisco. STP was actually synthesised by Sasha Shulgin a few years earlier (1963/1964) and given the name DOM (LINK). Chemically, DOM is 2,5-dimethoxy-4-methylamphetamine, the first member of a family of amphetamine derivatives (DOx) with the 2,5-dimethoxy pattern around the phenyl ring. All members of the DOx family also contain another group at the 4-position on the ring. The “M” stands for “methyl”, hence DOM. Other members include DOC, containing a chlorine at the 4-position; DOI, with an iodine; and the lesser know DOTFM, sporting a trifluoromethyl group (this one a Nichols creation).
DOM is around 100x more potent than mescaline (the prototypical benchmark psychedelic phenethylamine against which the potency of others are measured) with a standard dose being 3-10mg. To understand why it’s so potent, we need to consider three specific features of its structure:
The 2,5-dimethoxy ring pattern.
The alpha-methyl group.
The 4-methyl group.
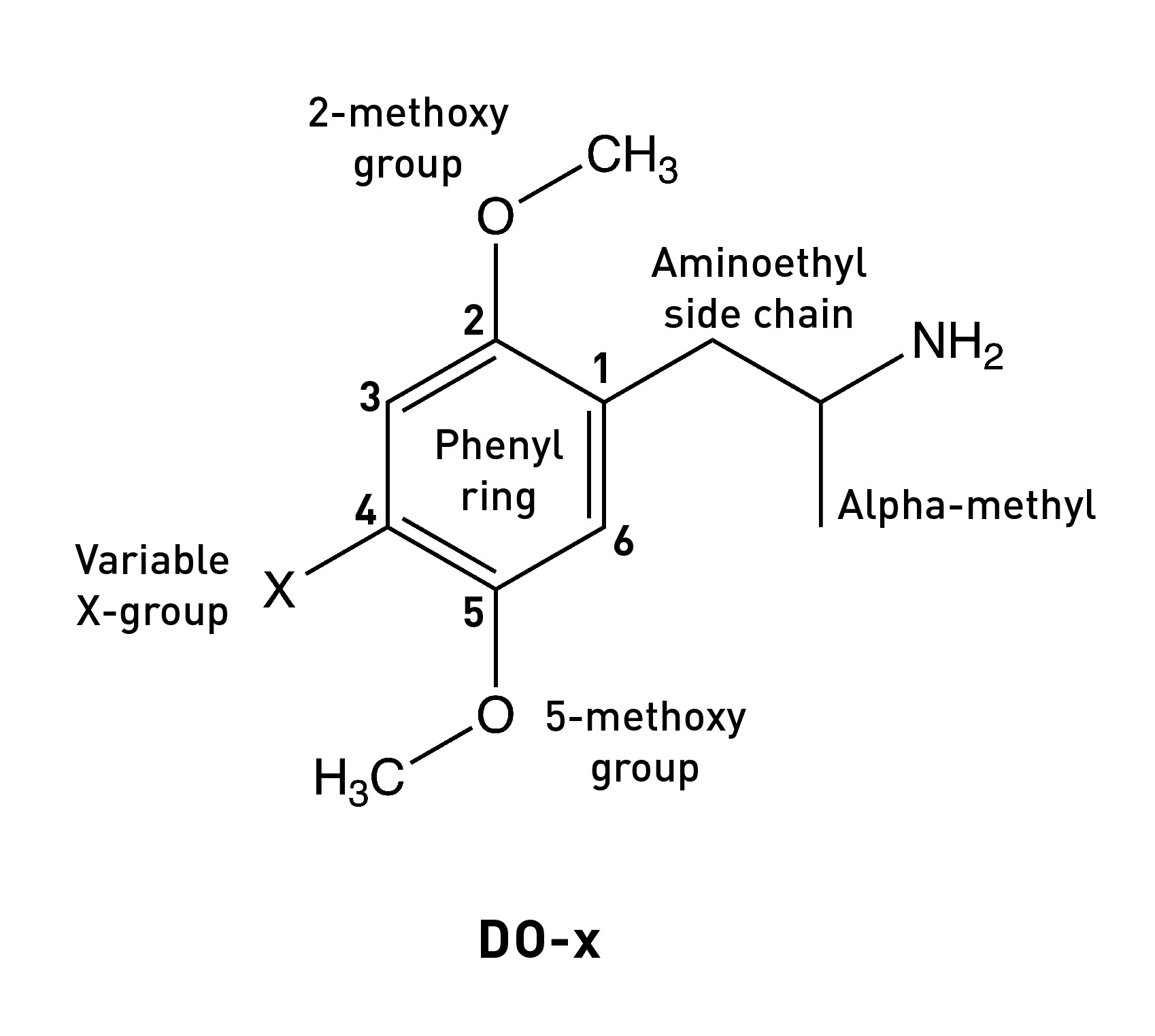
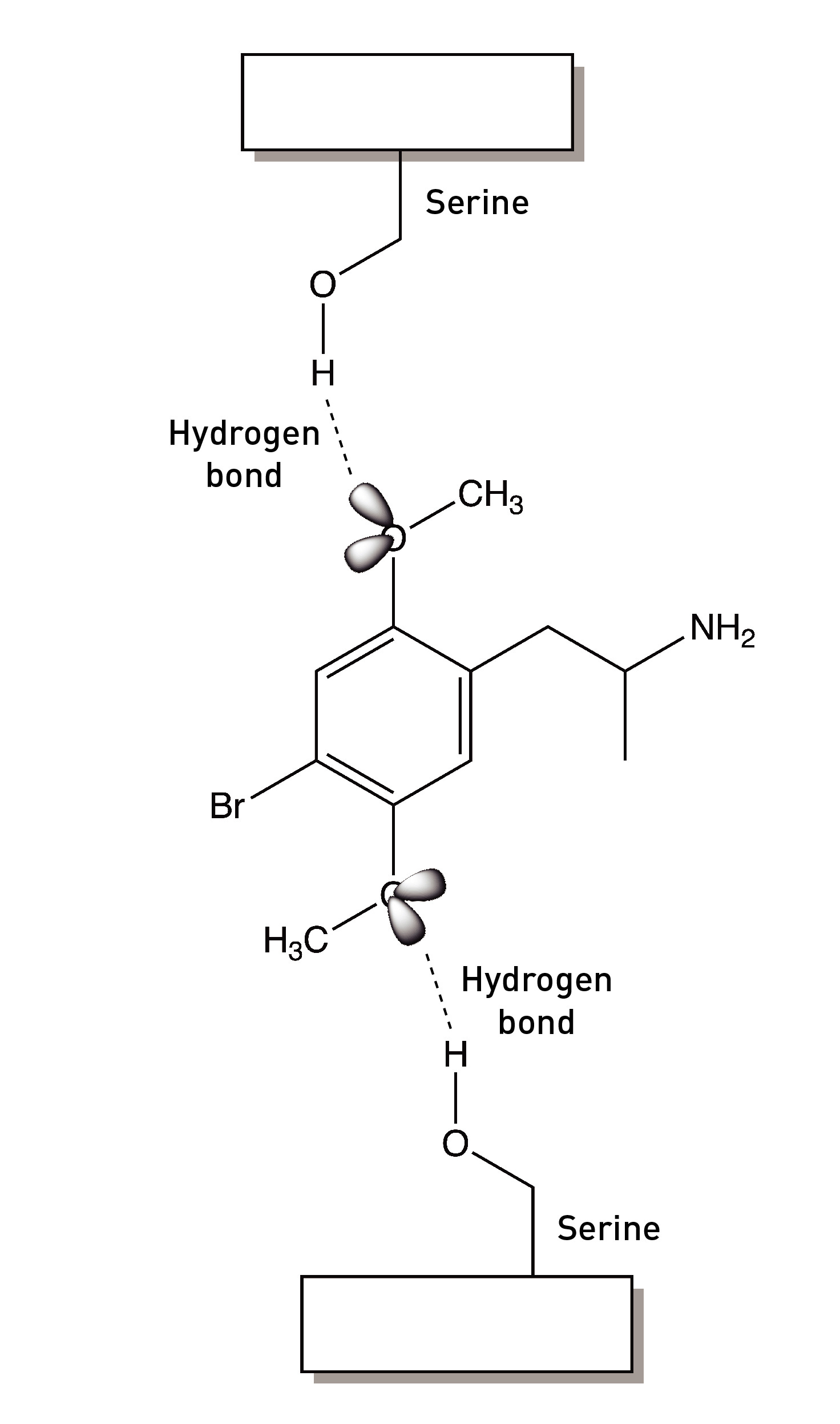
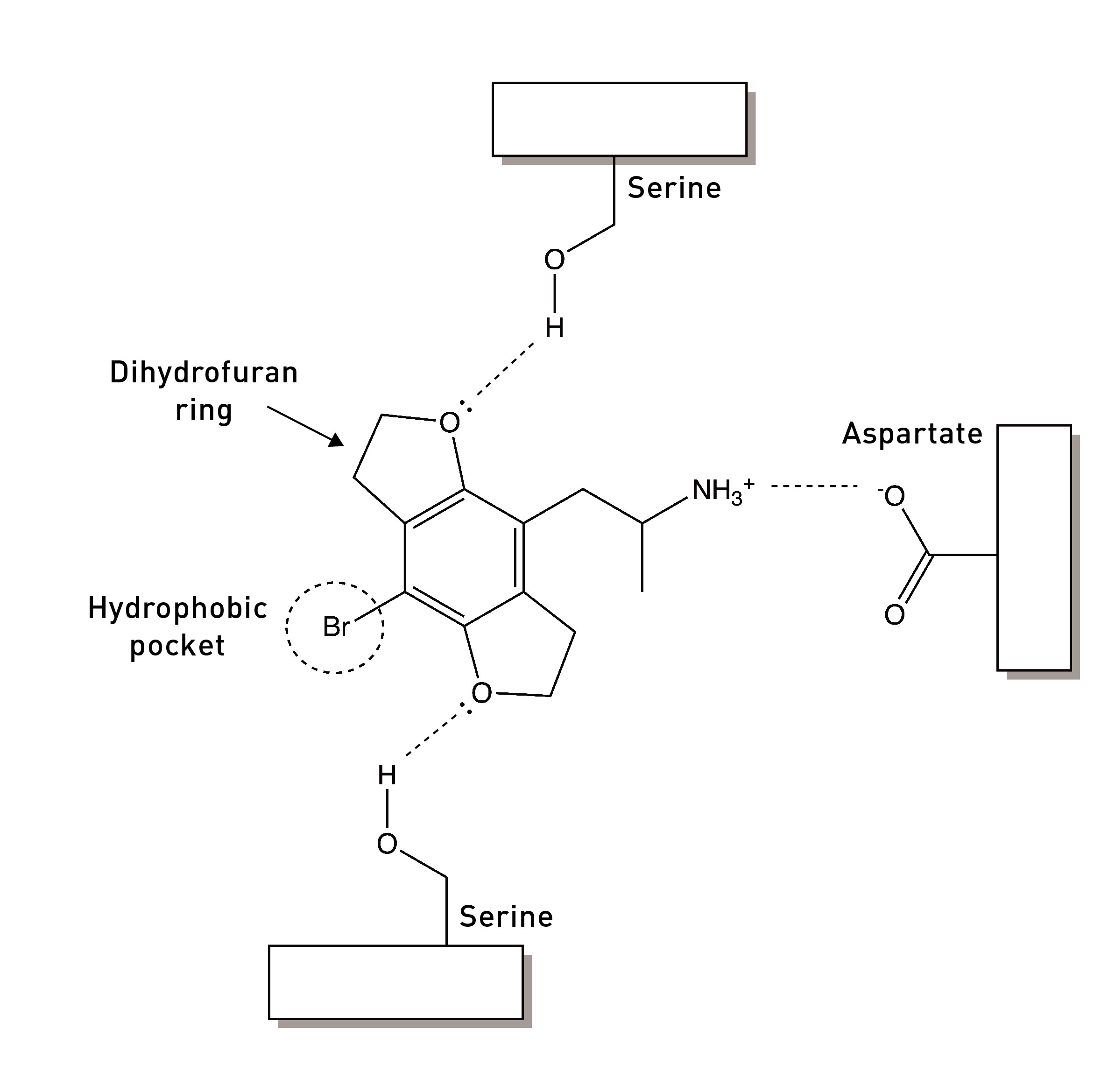
The two methoxy groups on the ring are essential for psychedelic activity and removing either abolishes the psychedelic effects. Molecular modelling studies show that the oxygens are well-positioned to form hydrogen binds with two serine amino acids within the 5HT2A receptor binding site (LINK). We’ll talk more about these interactions shortly, since they’re central to the development of Bromo-DragonFLY.
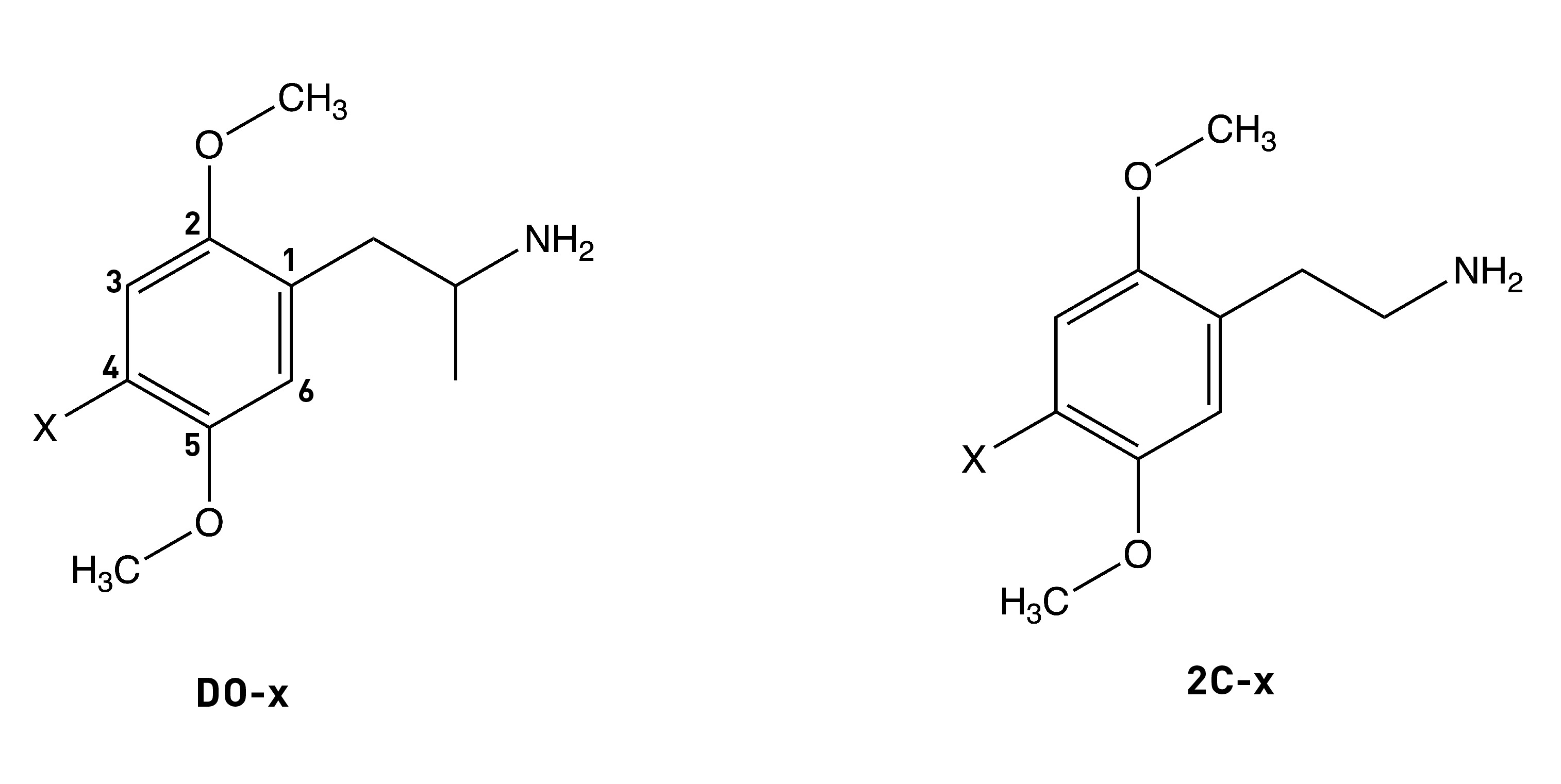
The alpha-methyl (i.e. next to the side chain amine) isn’t essential for psychedelic activity and removing it from any of the DOx drugs yields the corresponding 2Cx drug. In the case of DOM, the desmethyl (i.e. without the alpha-methyl) analogue is 2C-D, which is psychedelic in the 20-60mg range (compared to 3-10mg for DOM). A similar drop off in potency occurs in going from DOB to 2C-B or from DOI to 2C-I. Whilst the alpha-methyl group doesn’t increase the affinity of the molecule for the 5HT2A receptor, it does enhance the molecule’s ability to activate signalling through the receptor (i.e. its efficacy). The alpha-methyl might also protect the amine from removal during metabolism, as well as making the molecule overall more hydrophobic and thus better able to cross the blood-brain barrier.
The 4-methyl group of DOM is of particular significance, since changing this group can have dramatic effects on potency. Removing it entirely yields 2,5-dimethoxy-amphetamine (2,5-DMA), which isn’t particularly psychedelic, even at doses of 100mg+. Shulgin notes in PHIKAL that:
“since it appeared to be totally a physical trip with tremors and some cardiovascular push and nothing of a sensory nature, I chose to explore it no further.”
Since the methyl group is hydrophobic, it’s thought to form an interaction within a corresponding hydrophobic region of the 5HT2A receptor that’s important in binding and activation. And, indeed, changing the methyl group to an even more hydrophobic group yields an even more potent molecule: DOB (with a bromine instead of a methyl) is highly psychedelic in the 1-3mg range. A similarly potent analogue is DOI, with an iodine in the 4-position (active at 1.5-3mg). However, we seem to hit a ceiling of potency with DOB. So, where to go from here?
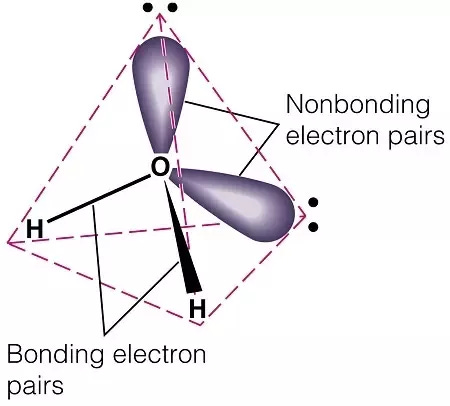
The Nichols group turned back to the methoxy groups (LINK). The oxygen atom of each methoxy group forms two covalent bonds: one to a carbon of the phenyl ring and the other to the methyl group carbon. It also contains two lone pairs of electrons not involved in covalent bonding to another atom. These bonding and non-bonding electrons repel each other and maximise their distance apart by adopting a tetrahedral arrangement around the atom. It’s these lone pairs that form a hydrogen bond to a well-positioned serine in the 5HT2A receptor binding site.
The bond connecting the methoxy group to the phenyl ring can rotate to generate two rotational conformers: one with the lone pairs pointing towards the side chain (the syn conformer) and the other with the lone pairs pointing away from the side chain (the anti conformer). Since there are two methoxy groups, we have four conformers in total: (2-syn, 5-syn), (2-syn, 5-anti), (2-anti, 5-syn), and (2-anti, 5-anti).
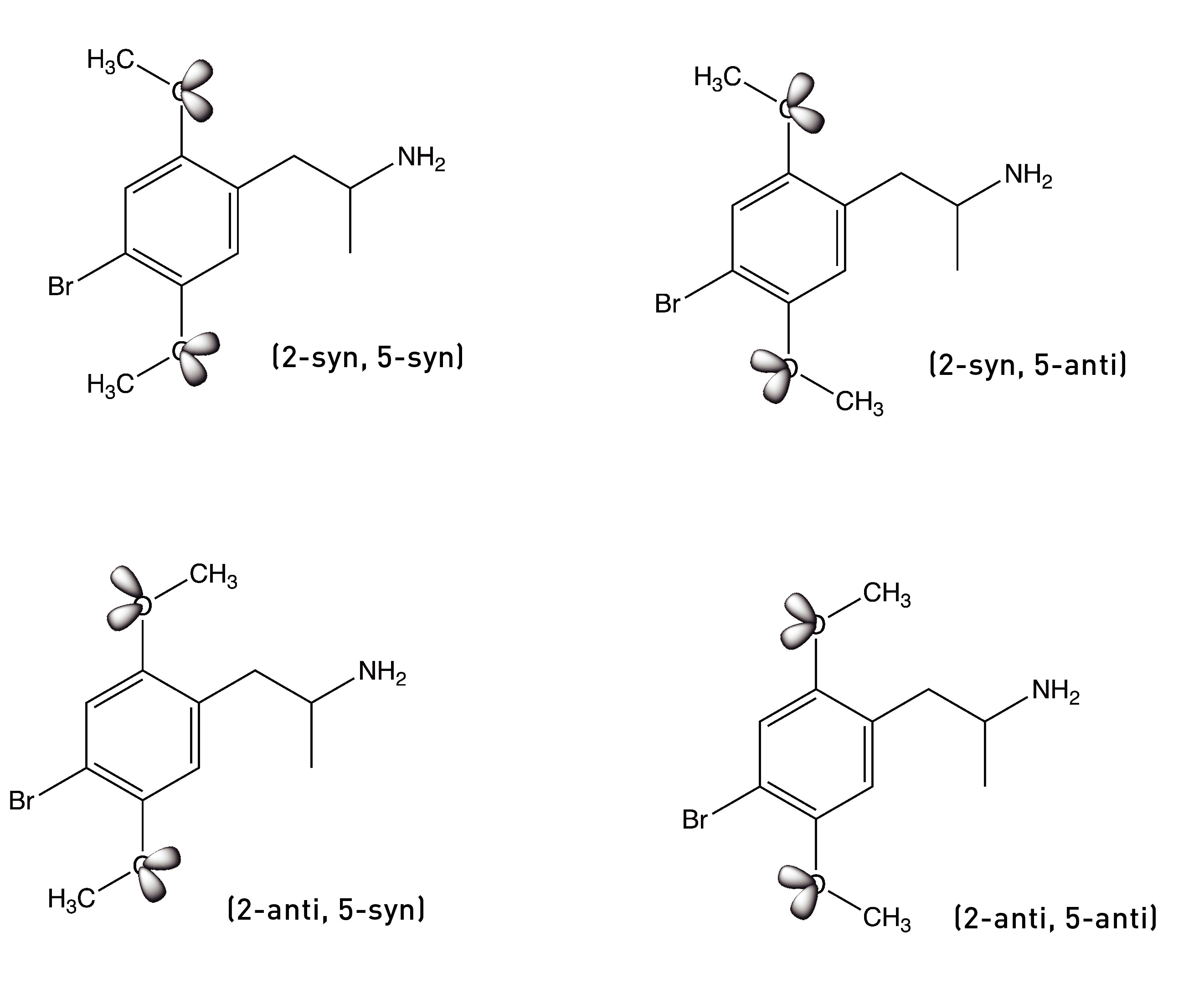
The Nichols group surmised that, as DOB binds to the 5HT2A receptor, each methoxy group must rotate into the conformer that brings the lone pairs closest to the serine to form the hydrogen bond. To test this idea, they built two molecules in which the oxygen at the 5-position was locked in either the anti or syn direction by a 5-membered ring — technically, a dihydrofuran ring. The molecule below on the left — with the lone pairs in the syn direction — showed much weaker activity compared to the parent compound, DOB. However, the molecule on the right — with the lone pairs in the anti direction — was equipotent with DOB. This suggested that the lone pairs must rotate into this anti direction to form the crucial hydrogen bond.
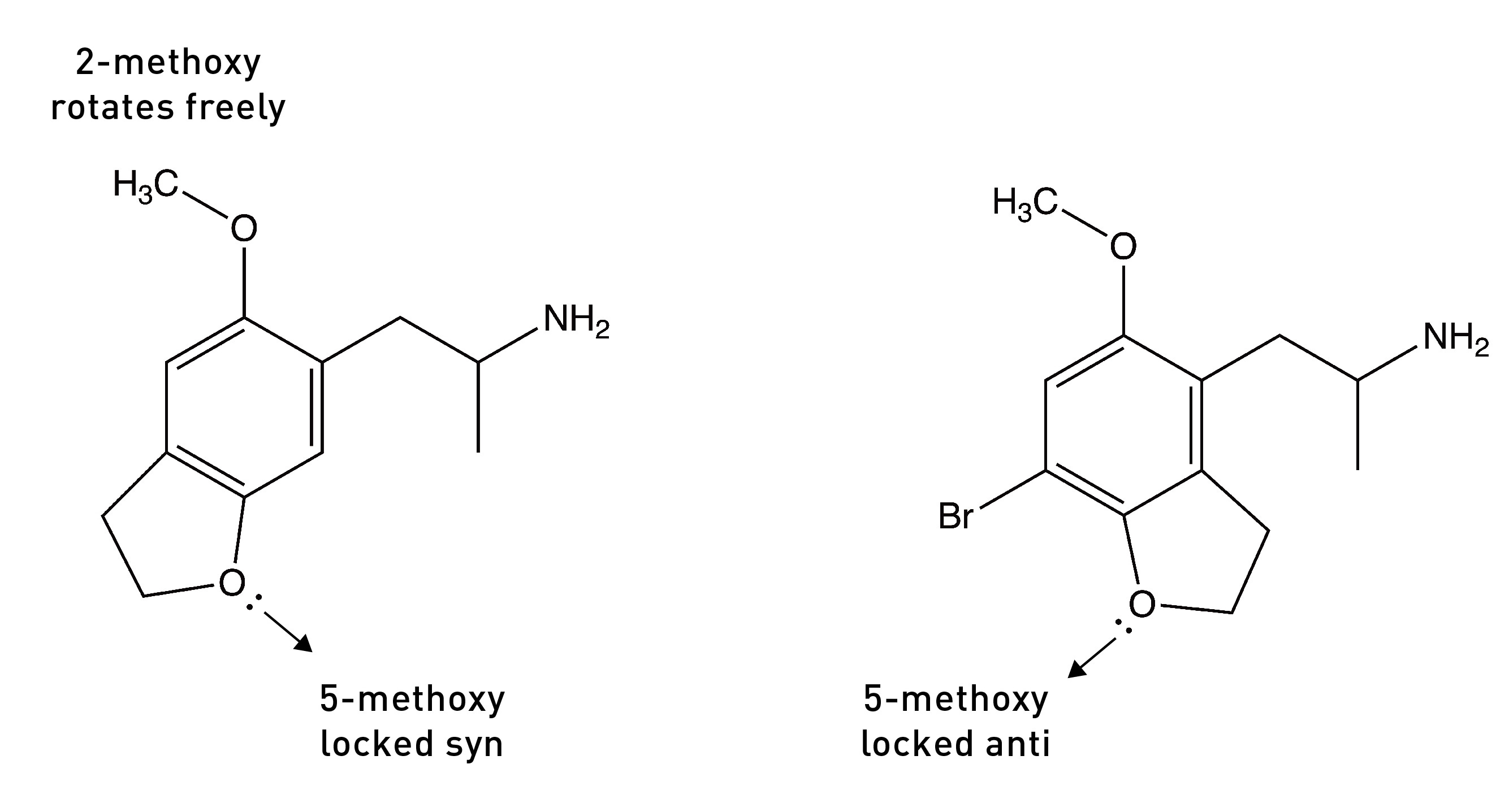
They had previously shown that a molecule in which the oxygen lone pairs at the 2-position were locked in the anti-conformation (pointing away from the side chain) by a 7-membered ring was a much weaker 5HT2A agonist than DOB, suggesting that the 2-position oxygen lone pairs must rotate into the opposite syn position to form its hydrogen bond with the other serine.
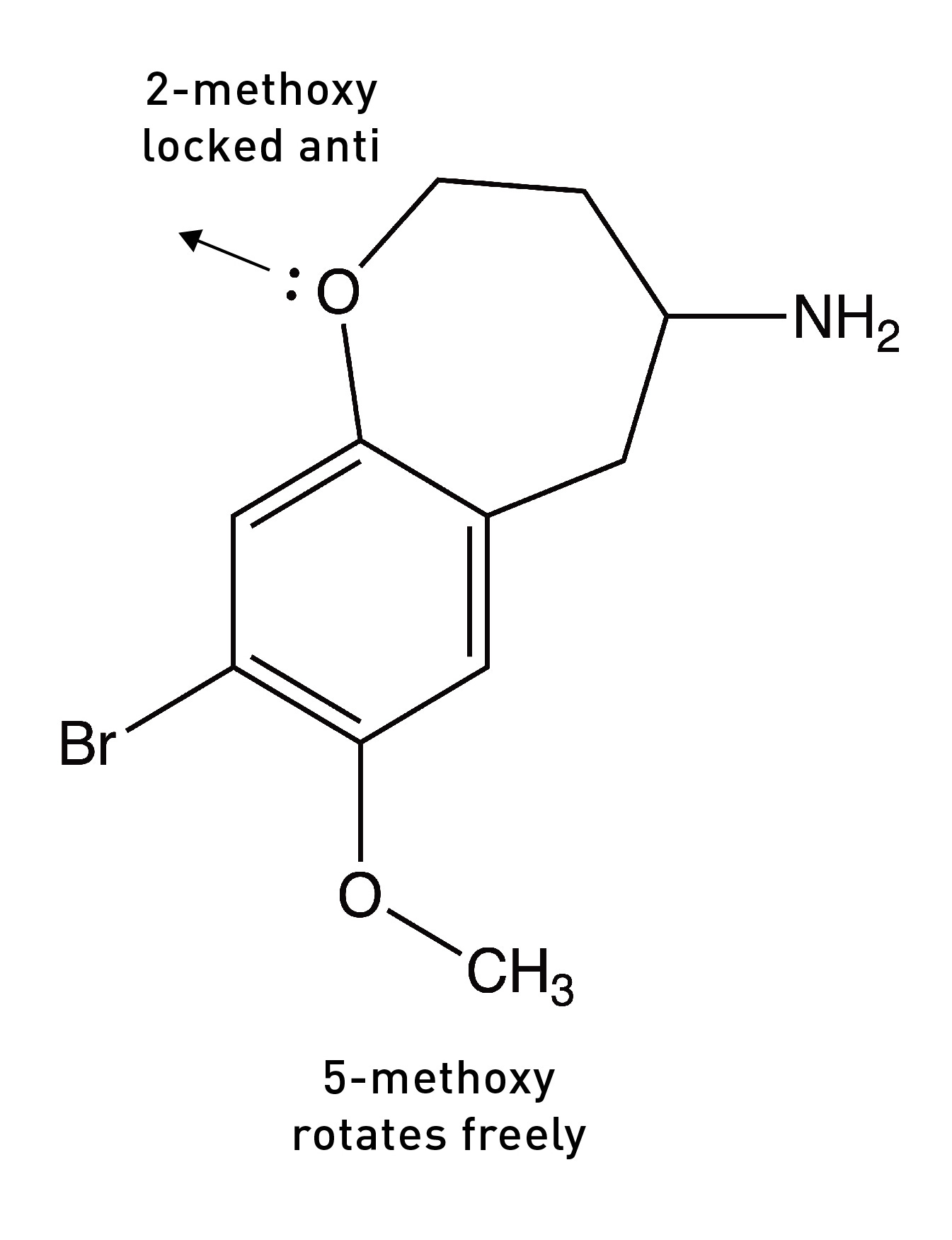
So, overall, the DOB likely binds to the 5HT2A receptor in the (2-syn, 5-anti) rotational conformation. Based on these results, they built a molecule in which both oxygens were locked into the “correct” rotational conformation. Remarkably, this molecule — known as DOB-FLY — was around 18x more potent than DOB and only about 50% less potent than LSD (in rats), making it the most potent psychedelic phenethylamine yet created. But they didn’t stop there.
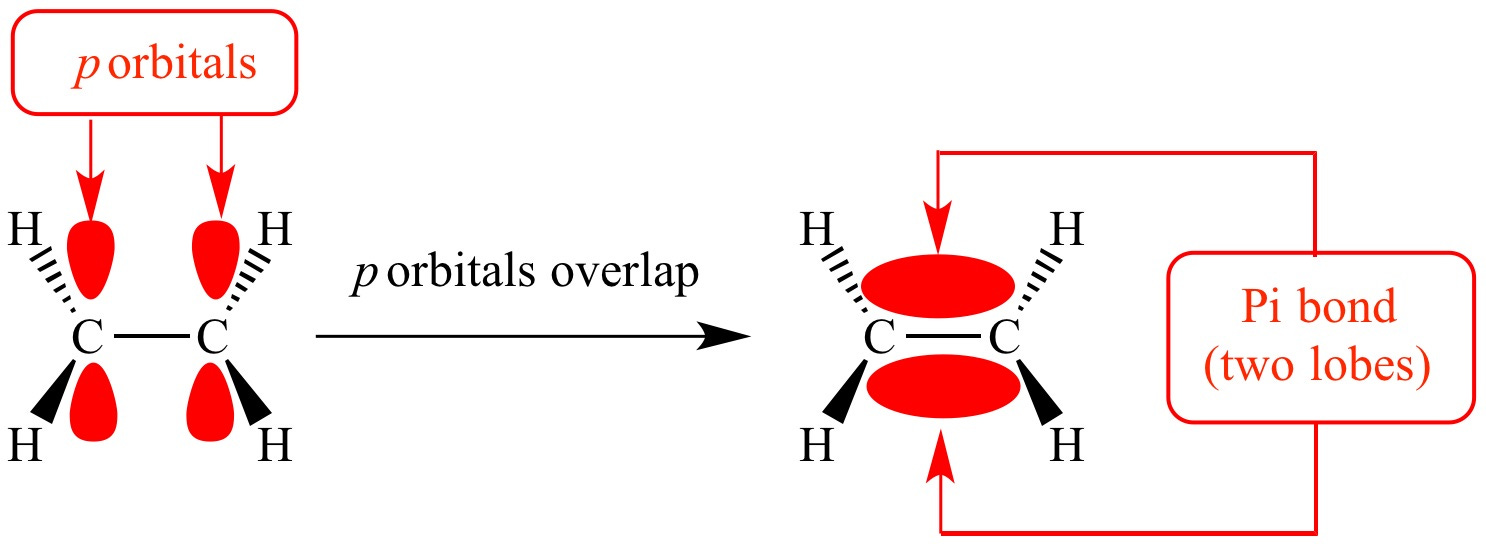
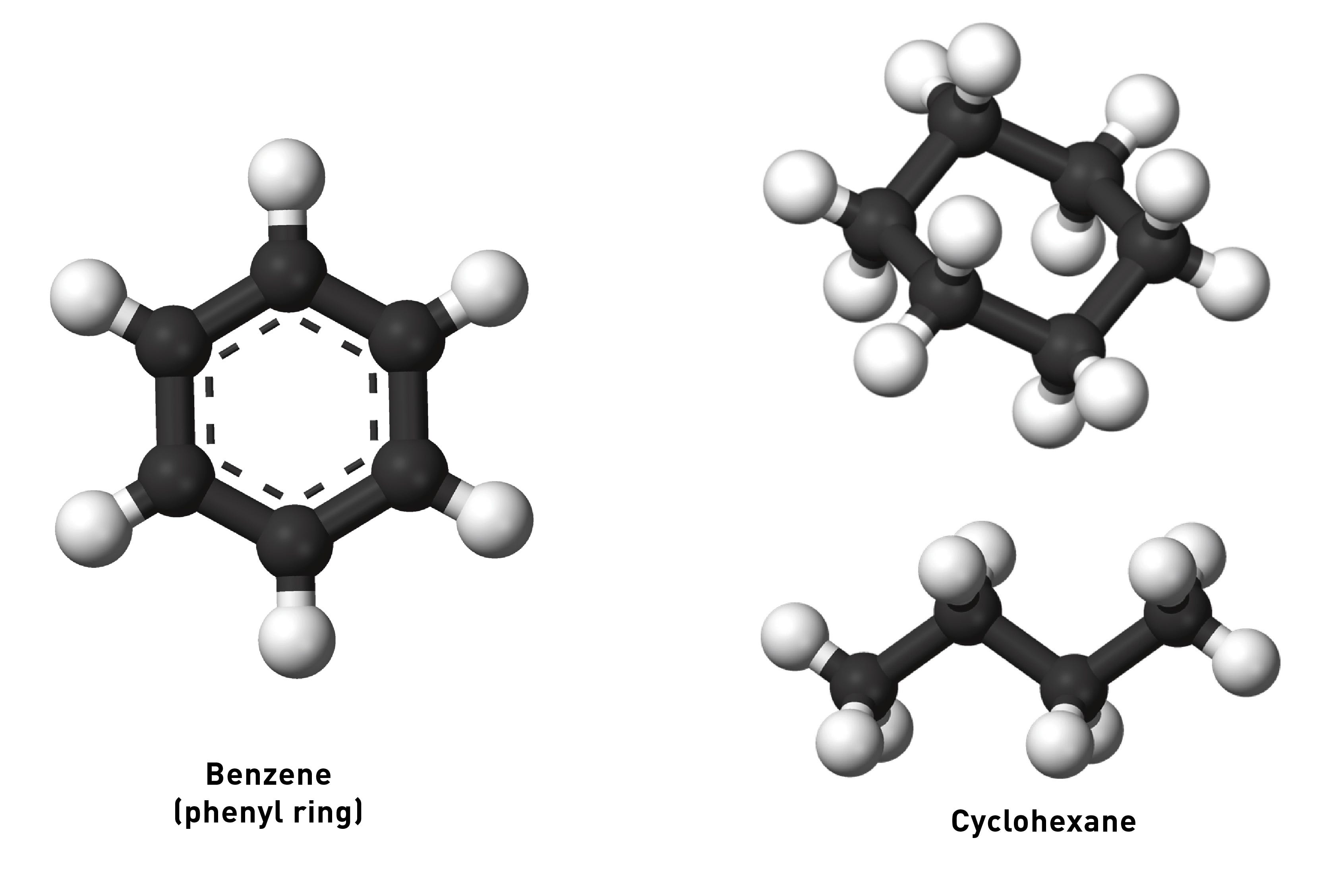
First, a bit of chemistry. The 6-membered phenyl ring of the basic phenethylamine skeleton has a perfectly flat hexagonal shape. This is because of the three carbon-carbon double bonds within the ring. When carbon forms a double bond to another carbon, the electrons that form the “second” bond sit in dumbbell-shaped p-orbitals that interact laterally (side-by-side) (these are known as pi-bonds). In fact, when there are precisely three double bonds in a 6-membered carbon ring, all of these p-orbital electrons interact and form a delocalised “cloud” of pi-bonded electrons above and below the ring. Molecules with these delocalised electrons are known as aromatic molecules (many of these molecules, but not all, have a distinct aroma, which is where the name originally came from).
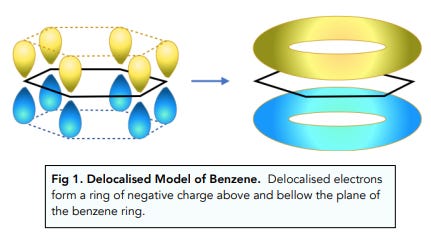
Compare the shape of benzene (with three double bonds) with cyclohexane (without any double bonds).
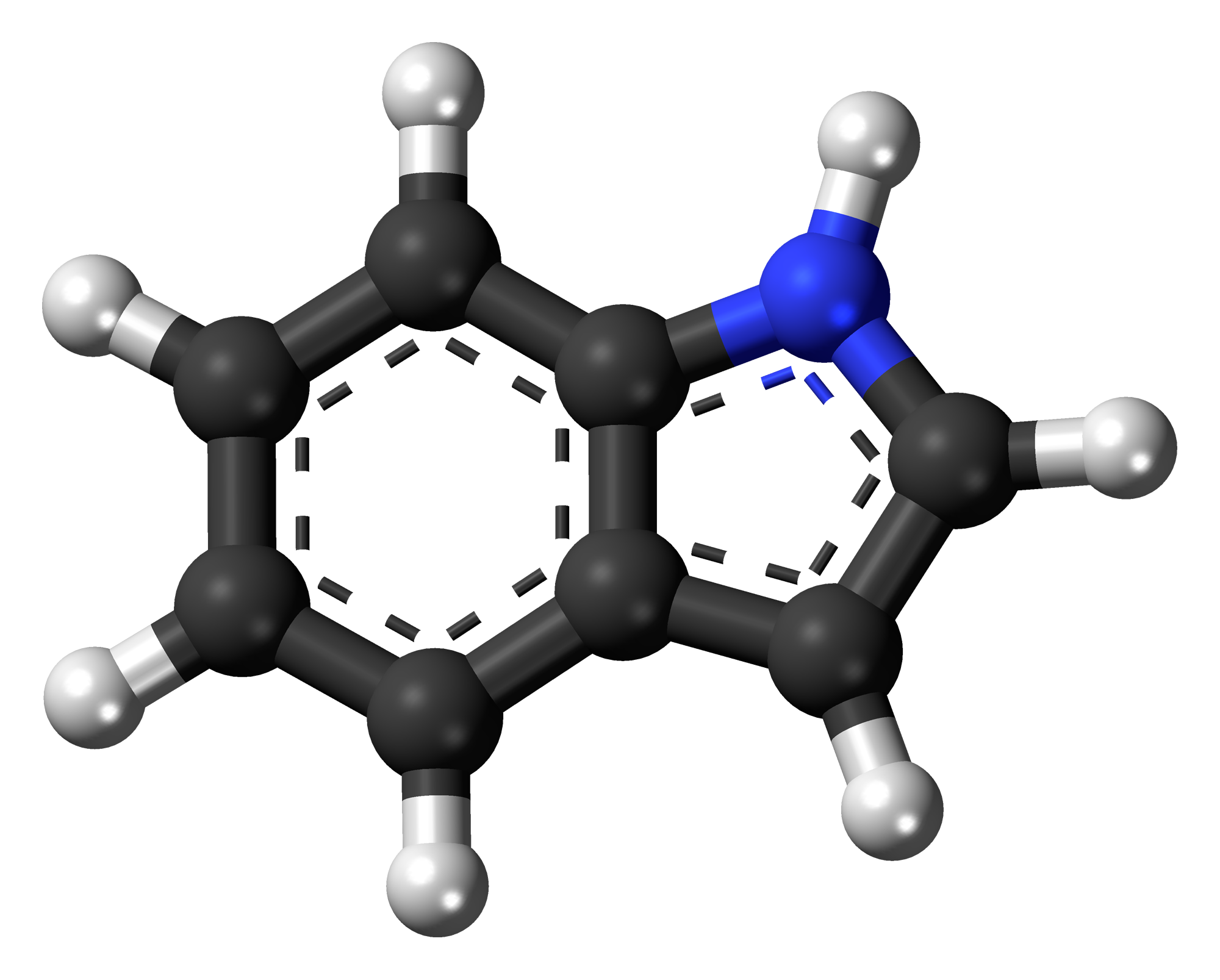
The indole system, which contains two connected rings, is also aromatic (and flat), since all of the p-orbital electrons can form this delocalised cloud (with the help of the lone pairs on the indole nitrogen atom):
This flat shape of aromatic molecules allows them to slide next to other aromatic amino acids in receptor proteins and form aromatic-aromatic (pi-pi) interactions that hold the molecule in place and contribute to receptor activation.
The Nichols group realised that they could easily convert the carbon-carbon single bond in the dihydrofuran ring of DOB-FLY into a double bond (forming a furan ring) (LINK). This would allow the furan ring electrons to interact with the pi-electron cloud of the phenyl ring, effectively creating an extended flat aromatic structure. Although this was something of an educated guess, it was surmised that this might enhance the aromatic-aromatic interactions within the 5HT2A receptor binding site that are known to form with the phenyl ring. This guess paid off, since they ended up creating a molecule even more potent (in rats at least) than LSD.
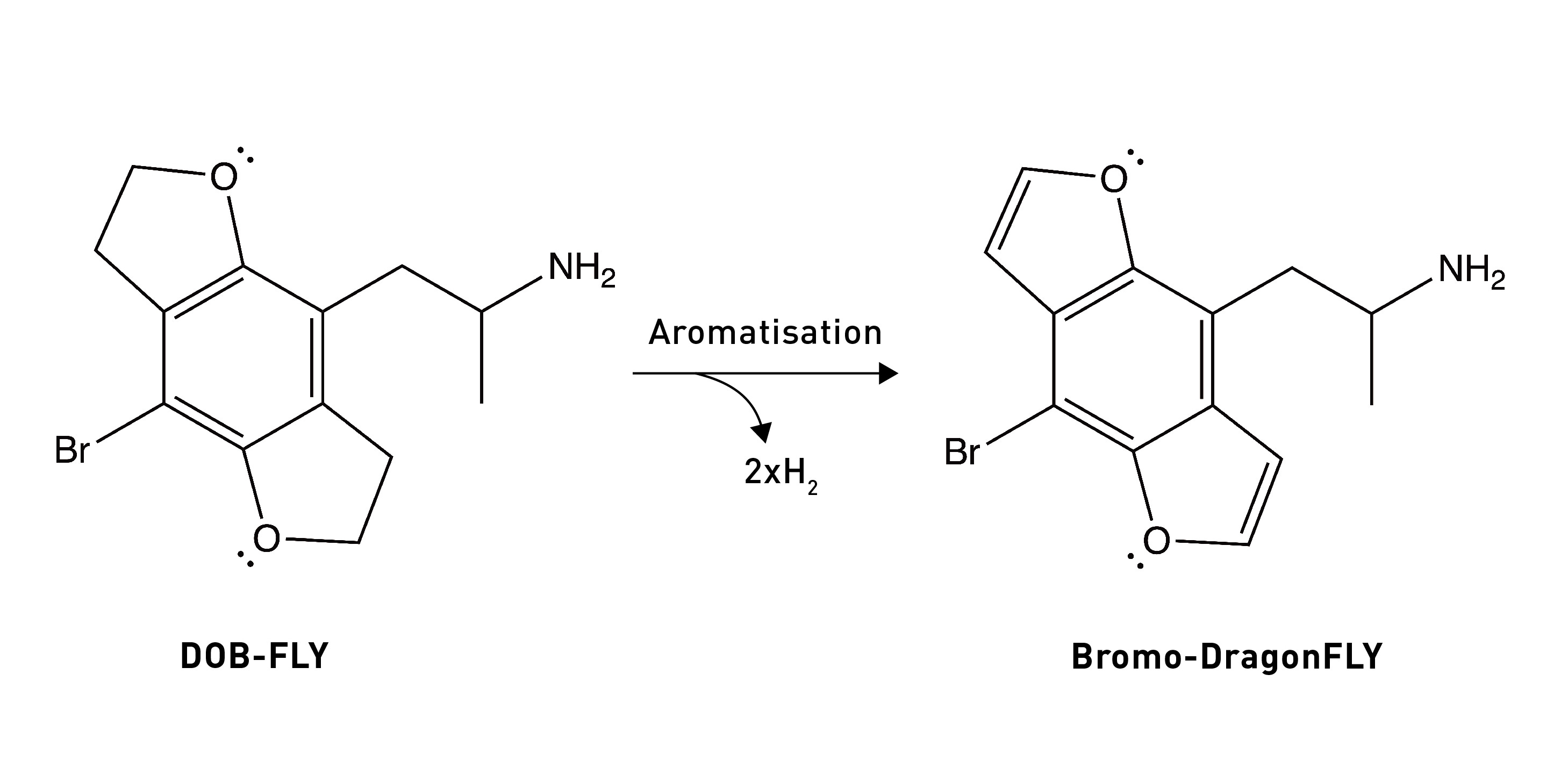
In humans, Bromo-DragonFLY is active in the 200-800+ microgram range, making it somewhat less potent than LSD (so, no, Bromo-DragonFLY is not the world’s most potent psychedelic), but still a mightily potent psychedelic indeed. However, Bromo-DragonFLY beats LSD by quite a margin when it comes to effect duration, with some users describing trips lasting days.
As I discussed in the last post, LSD’s 6-12+ hour trip can be largely explained by the unique way it interacts with the 5HT2A receptor. Bromo-DragonFLY’s extremely long trip has an entirely different explanation. When researchers incubated Bromo-DragonFLY with several of the most common metabolic enzymes found in the liver, they found it to be perfectly stable and resistant to metabolic transformation. Its close cousin 2C-B-FLY, which doesn’t contain the extended aromatic system, was rapidly converted to inactive metabolites (LINK).
Bromo-DragonFLY was also found to bind strongly to proteins in blood plasma, which likely further restricts its availability for enzymatic degradation and elimination. After ingestion, these plasma proteins soak up the molecule and very gradually release it into the bloodstream — like an in-built slow-release mechanism. Coupled with the sluggish metabolism and excretion, this makes for an extremely extended trip.
But it’s not merely the mercilessly long trip users should be concerned about. Unlike LSD, which is remarkably benign, even at heroic doses, Bromo-DragonFLY can produce some pretty horrifying side-effects. In high doses, Bromo-DragonFLY can cause vasoconstriction (leading to necrosis and amputation in at least one case: LINK), seizures and, rarely, death (LINK). Sadly, Bromo-DragonFLY is occasionally sold on blotters as LSD, or some sellers/users simply confuse it with 2C-B-FLY and unwittingly consume a massive overdose (10mg is a standard dose of 2C-B-FLY — this would be a potentially fatal overdose of Bromo-DragonFLY). This certainly isn’t a psychedelic to be played with and again highlights the importance of testing your drugs. If you’re not 100% sure of what you’re taking, don’t take it.
Similar question. How could one make lsd last longer. With the onset of analogs this could be n interesting.
I am an undergraduate genetics major that has taken Gen Chem I, II, Orgo I, II, and Biochemistry, and I am in a pharmacogenomics class where I learned the basics of pharmacology, so it is cool to see you neatly tie everything together in posts about psychedelics! Also, quick question on the bromine. I believe in the article you said that bromine was included because it is (more hydrophobic?) than the methyl group. My understanding was that bromine was polarizable and could induce a dipole thus making it more hydrophilic as opposed to hydrophobic: what am I missing? Thank you very much, this post has made my plane ride much better!