



Dropping the NBOMe…
The anatomy of a super-potent psychedelic drug family

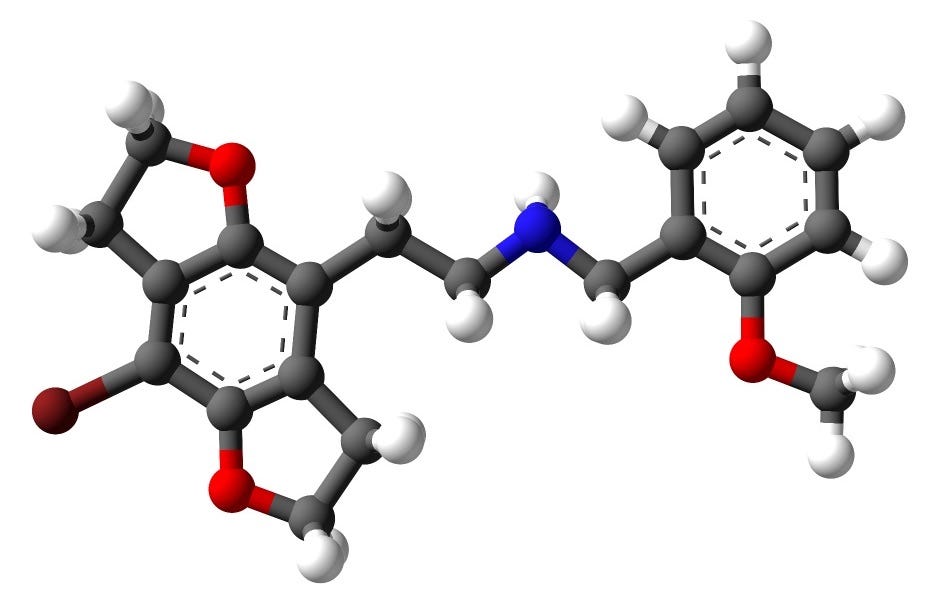
Whenever I’m confronted by a molecule with psychoactive properties, there are basically two questions I ask myself:
What is it about this molecule’s structure that makes it psychoactive?
How might its structure be tweaked to either enhance or alter its psychoactivity?
One of the most important concepts in medicinal chemistry is the pharmacophore, which describes the structural and electronic features of a molecule that are essential for its biological effects. According to IUPAC (International Union of Pure and Applied Chemistry), a pharmacophore is “an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger (or block) its biological response”.
So, a pharmacophore isn’t merely a property of the molecule itself, but rather a mapping between the molecule and a particular biological target. When talking about the classic psychedelics, we might ask: What features of a molecule are important in its binding and activation of the 5HT2A receptor?
Below is one way to depict the pharmacophore of molecules that interact with the 5HT2A receptor (don’t worry too much about the details — simply note how different features of the molecule are linked to specific interactions within the receptor binding site):
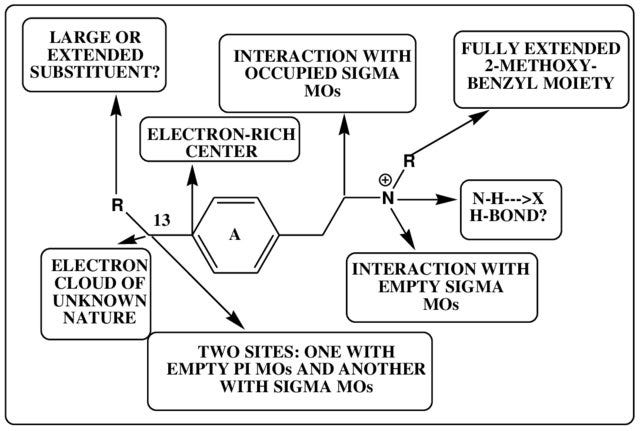
A pharmacophore is best thought of as a working model that can be used to inform the development of new receptor agonists and antagonists: based on our understanding of a molecule’s interaction with a receptor, which parts of the molecule can we change in the hope of creating a molecule with enhanced potency as an agonist and which should probably be left well alone to avoid diminishing it?
A pharmacophore model can rarely be said to be finalised and complete — there’s always the possibility of finding new ways of enhancing the interaction between a molecule and a receptor, perhaps by discovering alternate areas of the binding site with which a molecule can interact or even entirely novel modes of binding to elicit the conformational changes that underpin receptor activation. Even at the level of single receptors, chemical pharmacology is complicated.
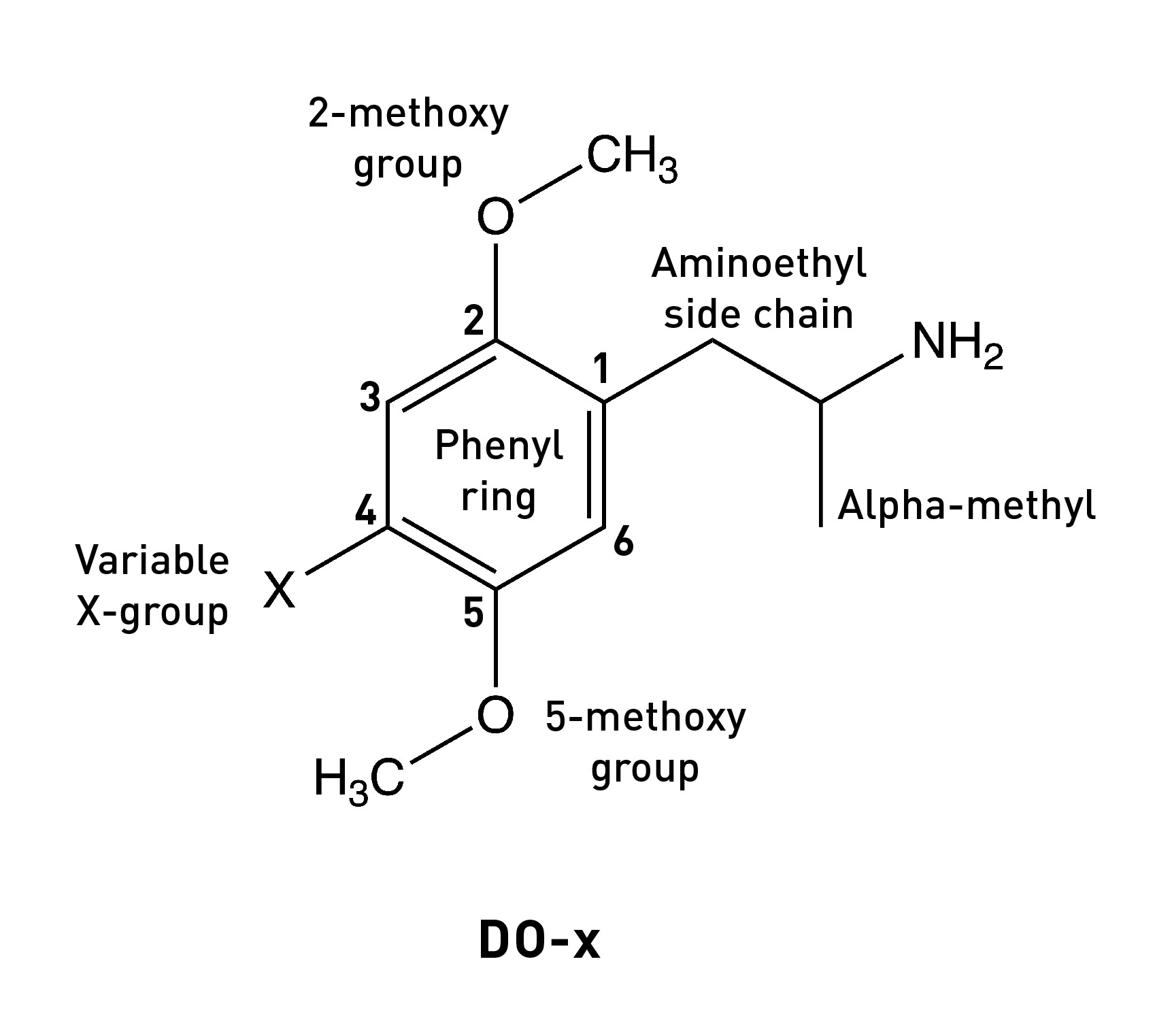
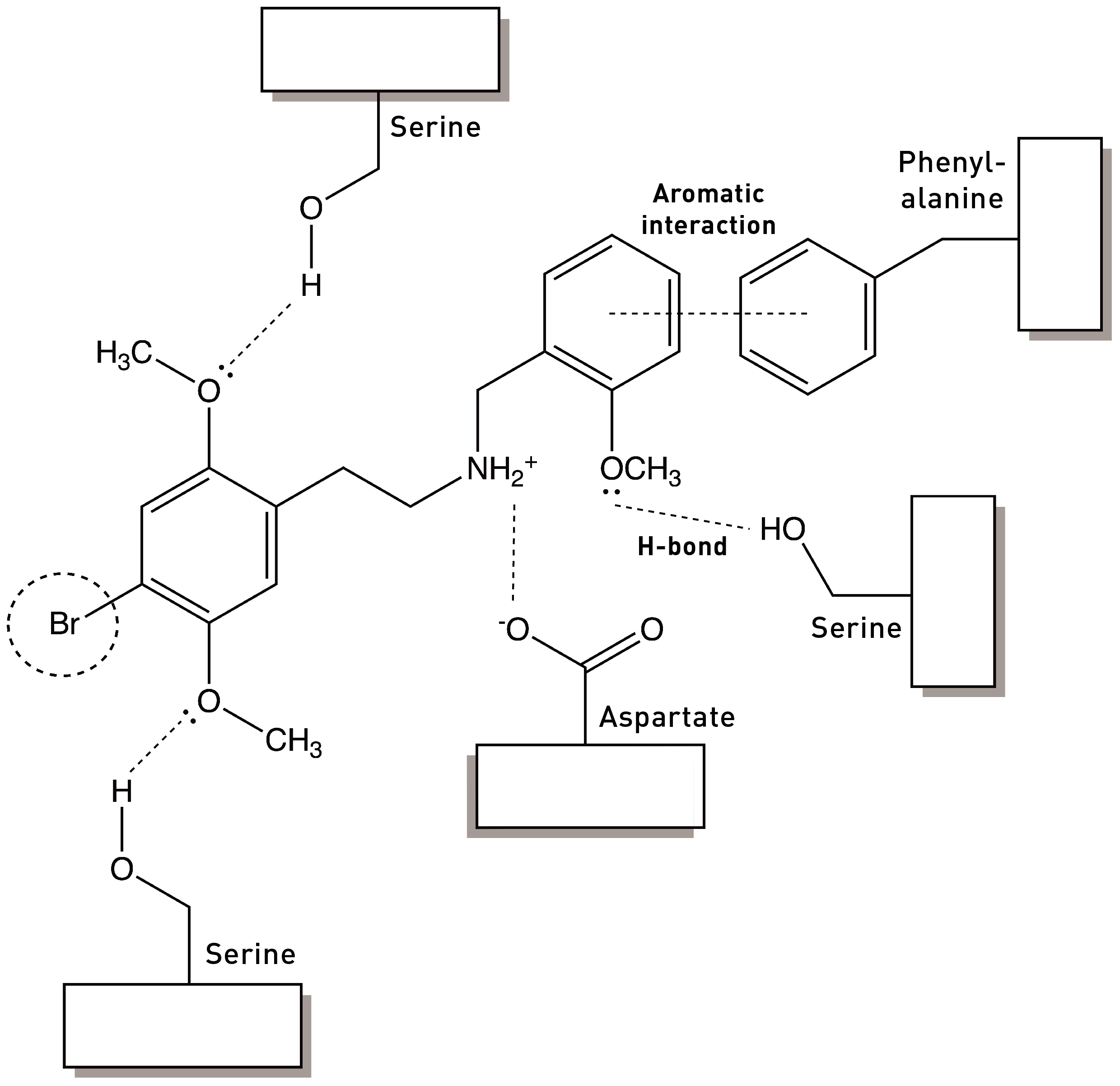
In the last post (please read if you haven’t already: HERE), I discussed how the 2,5-methoxy substituents of the DOx family of psychedelic phenethylamines are essential in binding and activating the 5HT2A receptor by forming hydrogen bonds with well-placed serines within the binding site — we might say these methoxy groups form part of the pharmacophore of the DOx molecules. Another important feature was the hydrophobic group at the 4-position on the phenyl ring, thought to interact with a hydrophobic pocket within the binding site. However, we’ve yet to consider perhaps the most important part of the pharmacophore of any molecule that binds and activates the 5HT2A receptor: the aminoethyl side chain amine.
The nitrogen of this amine forms an ionic bond with an aspartate amino acid that’s critical for binding to the 5HT2A receptor (LINK):
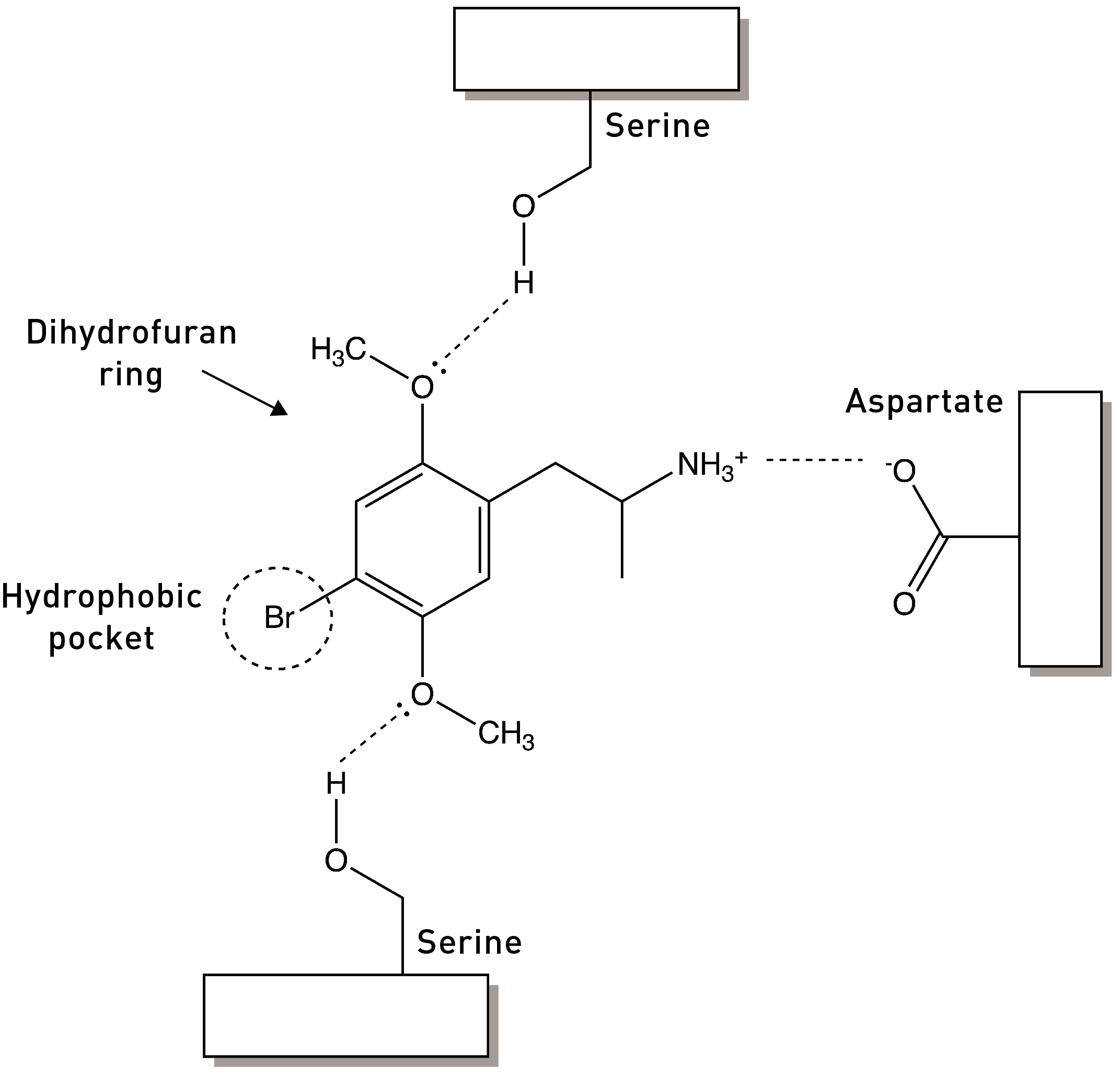
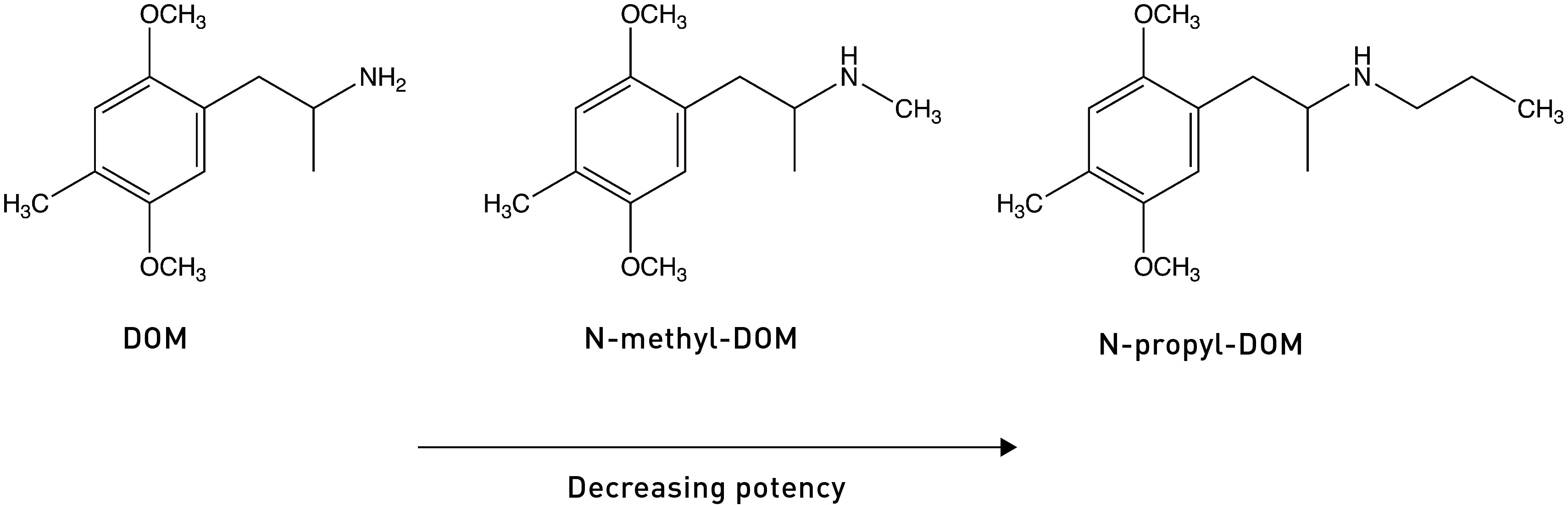
Adding alkyl chains — methyl, ethyl, propyl groups — to this amine dramatically diminishes the affinity and potency of the agonist. For example, N-methyl-DOM is nine times less potent than DOM, and N-propyl-DOM has a 30x lower affinity for 5HT2A receptors. This isn’t too surprising, since these increasingly bulky groups might prevent the amine nitrogen from getting close enough to that key aspartate to form an interaction, with detrimental affects on the molecule’s ability to bind and activate the receptor.
For many, this might be the point at which playing with groups on the amine might be considered a dead end. However, Richard Glennon of Virginia Commonwealth University published a paper way back in 1994 showing that adding a benzyl group — a phenyl ring plus a single carbon — to 2C-B (generating 25B-NB) actually increased its binding affinity for the 5HT2A receptor. A similar result can also be obtained by adding a benzyl to 2C-I (to generate 25I-NB) (LINK).
This is, at first glance, rather surprising, since a benzyl group is certainly bulkier than a propyl chain. So what’s going on?
Whenever adding a phenyl ring to a molecule enhances its binding to a receptor, “aromatic-aromatic interactions” should immediately spring to mind (see my last post): It’s likely that the phenyl ring is forming an interaction with one of the three amino acids containing delocalised aromatic ring structures: phenylalanine (containing a phenyl), tyrosine (containing a phenol — “hydroxy-phenyl”), or tryptophan (with an indole ring). In the case of the 5HT2A receptor, there’s a particular phenylalanine in the binding site that’s known to play a role in receptor activation and which is well-placed to interact with the benzyl group in 25B-NB. Indeed, if this phenylalanine is mutated (changed to another amino acid without the phenyl ring), the affinity of 25B-NB drops dramatically. So, by attaching this benzyl group, we have added an entirely new interaction within the 5HT2A receptor binding site (rather than merely modifying existing interactions).
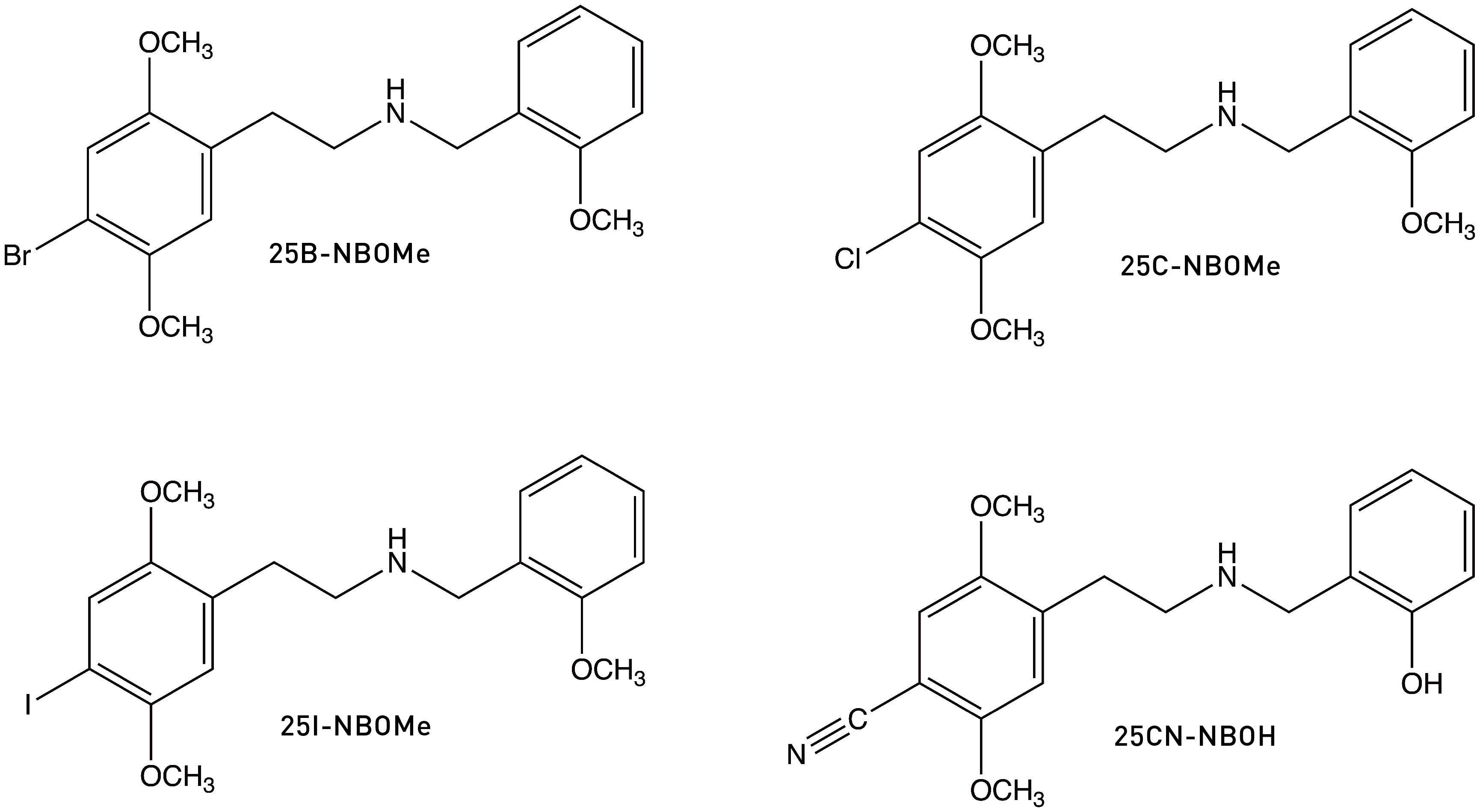
Now, once we have this phenyl ring in place, it’s natural to wonder whether receptor binding can be enhanced even further by adding groups to the ring. Dave Nichols and his team duly wondered and then set about building such molecules. Most notably, they found that adding an oxygen-bearing substituent to the 2-position of the ring greatly increased 25I-NB’s affinity for the 5HT2A receptor. Specifically, adding a hydroxyl group — generating 25I-NBOH — increased the affinity almost 3-fold. Adding a methoxy group — generating 25I-NBOMe — increased it almost 4-fold, producing a molecule only barely less potent than LSD, with an ED50 (in rats) of 78 μg/kg vs 53 μg/kg LSD (LINK).
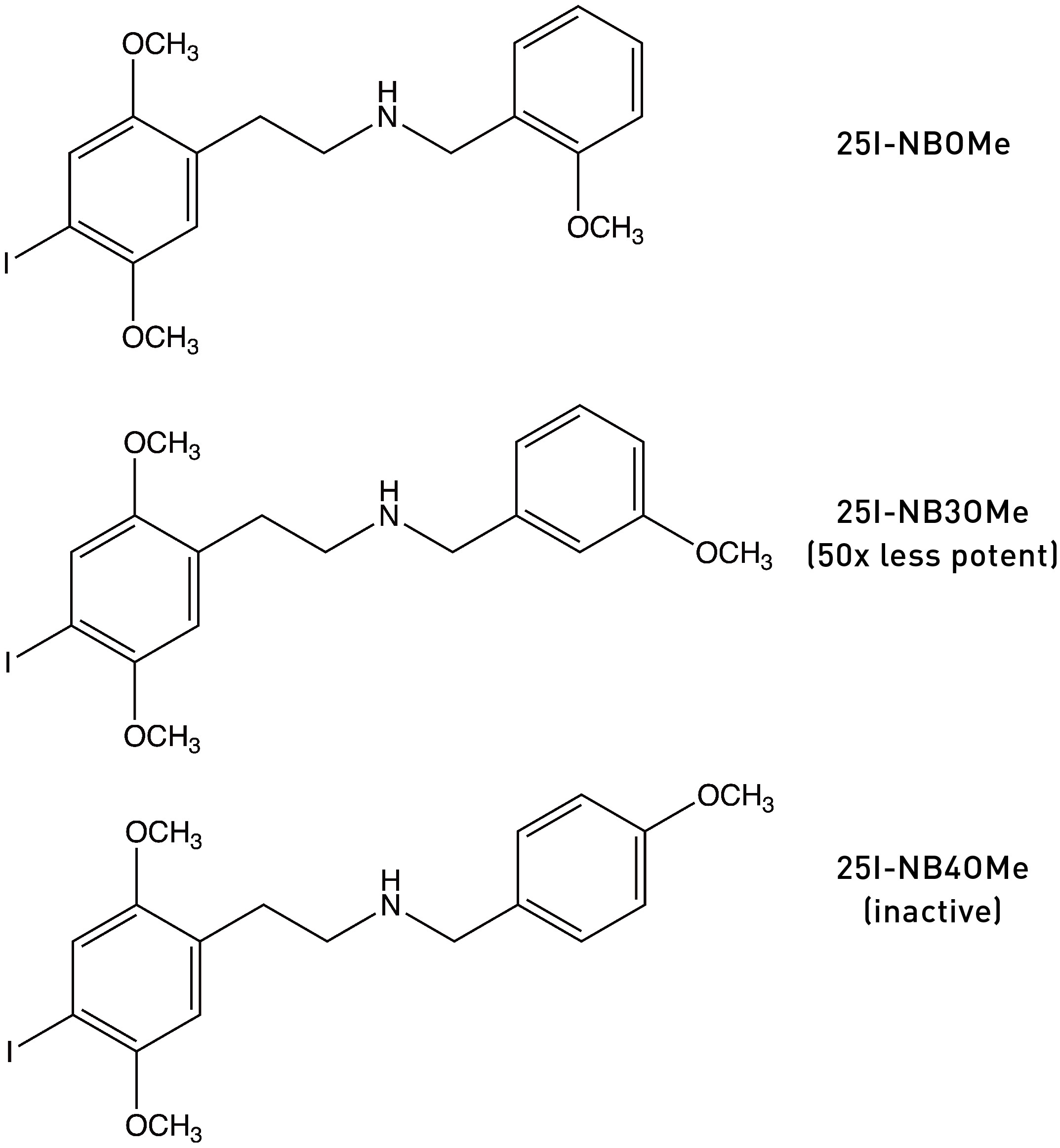
The most obvious explanation for the increase in binding affinity and potency when adding an oxygen-bearing substituent to a molecule is the formation of a hydrogen bond within the receptor binding site. In support of this hypothesis, moving the methoxy group of 25I-NBOMe to the 3-position or 4-position progressively reduces both receptor affinity and potency: 25I-NB3OMe is 50x less potent than 25I-NBOMe, and 25I-NB4OMe is completely inactive (LINK). This makes sense, since formation of a hydrogen bond requires the oxygen lone pair to be positioned close to the H-bond acceptor in the binding site, so a methoxy in the wrong ring position is not only unable to form the hydrogen bind but might also block receptor binding completely.
In 2020, Bryan Roth’s group published a crystal structure of the 5HT2A receptor bound to 25CN-NBOH, which demonstrated formation of this hydrogen bound with a binding site serine (LINK), as well as the aromatic interaction between the benzyl group and a phenylalanine as hypothesised. So, this gives us a more complete picture of the NBOMe/NBOH interactions with the 5HT2A receptor:
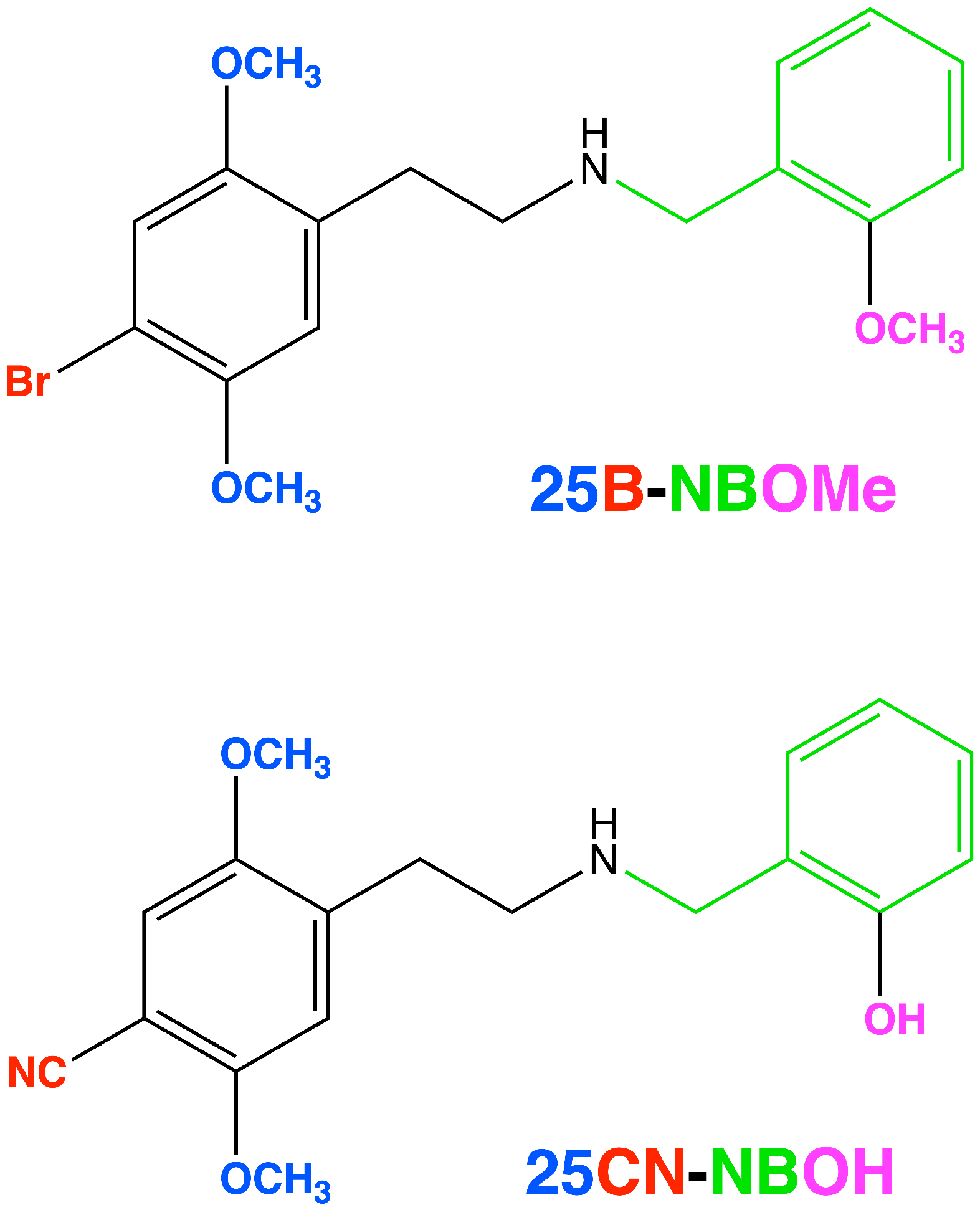
A note about naming: "25” refers to the 2,5-methoxy pattern on the main phenyl ring, with the following letter (as in 25B or 25I) referring to the 4-position substituent. The “NB” stands for “N-benzyl”, with the final “OH” or “OMe” referring to the hydroxyl or methoxy, respectively. So, these molecules are known collectively as the 25X-NBOx family. 25I-NBOH, 25CN-NBOH, 25B-NBOMe, 25C-NBOMe, 25I-NBOMe are some of the most well-known representatives, but the total number of analogues that have been synthesised and explored to varying degrees approaches (and might now exceed) 100 different molecules.
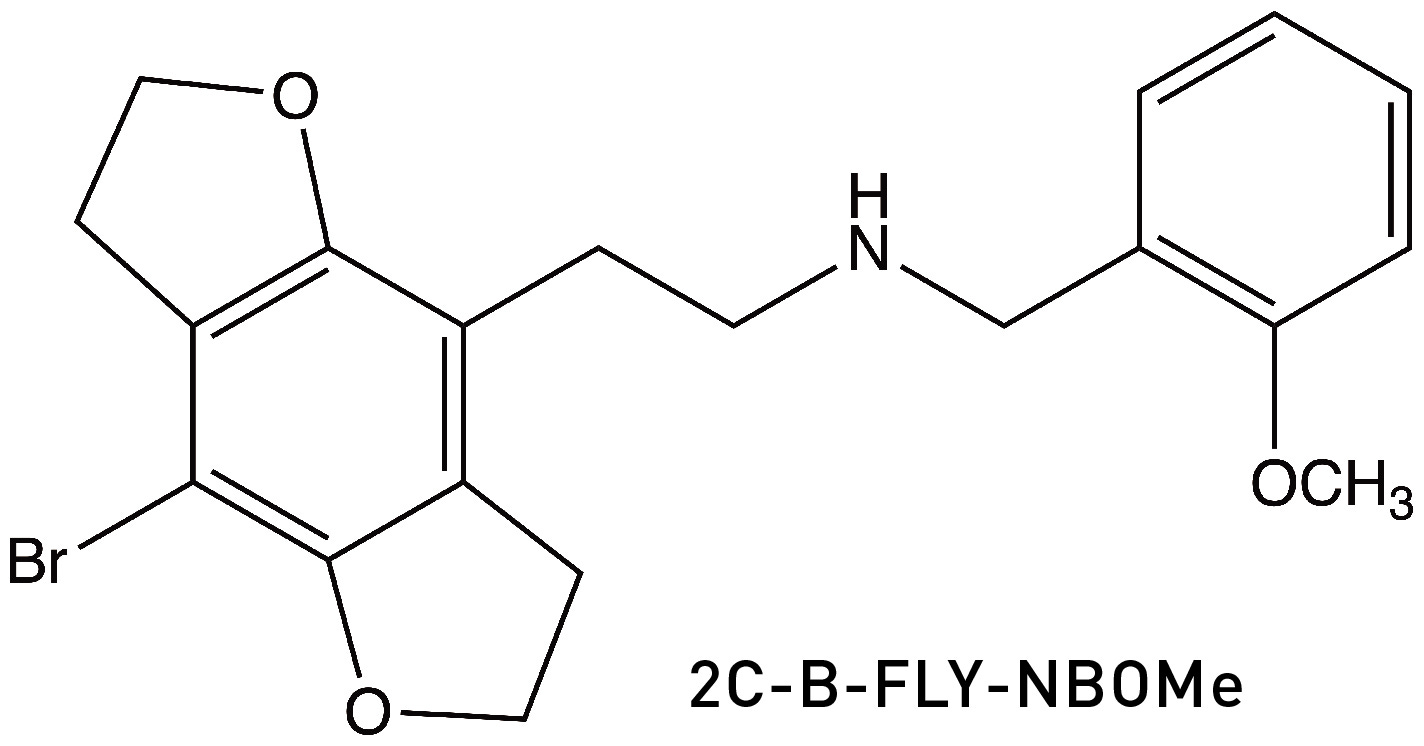
If you’re wondering whether anyone has tried adding a methoxy-benzyl group to one of Nichols’ FLY molecules in the hope of generating an even more potent psychedelic, indeed this has been synthesised (LINK):
The scant reports of human use of 2C-B-FLY-NBOMe indicate activity in the 100-200 microgram range, which is similar to the other NBOMe molecules. It feels like we’re pushing the limit of potency achievable by manipulating the structure of psychedelic phenethylamines. So, quite remarkably, LSD, a semi-synthetic psychedelic discovered by accident over 80 years ago, still maintains its record as the most potent psychedelic known, despite the efforts of some of the best medicinal chemistry minds in the world. That’s not to say that the NBOMes/NBOHs aren’t super potent — they most certainly are — but the potency ceiling of LSD appears to be a formidable one to crack.
So, are the NBOMes and NBOHs worth the trip? Well, there are a lot to choose from, many of which have almost no reported human use, so we can only speak about the handful that have become reasonably popular. As with all psychedelics, effects can vary wildly between individuals, but most reports describe the effects as being broadly comparable to LSD, but this is not a particularly helpful descriptor. However, the NBOMes and NBOHs are sometimes sold as “synthetic LSD” or even mis-sold as actual LSD (with potentially fatal consequences). Effects generally last 6-8 hours and users report NBOHs such as 25I-NBOH as being slightly less potent than the corresponding NBOMe and with a shorter duration.
Sublingually, the threshold for the onset of hallucinogenic effects is reportedly about 100–250 μg, with mild effects at 250–450, strong effects at 450–800, and very strong effects over 800 μg. Owing to their potency, it’s very easy to unwittingly enter into overdose territory with the NBOMes/NBOHs and, unlike LSD, this is accompanied by very real physiological dangers: hyperthermia, protracted seizures, intracerebral haemorrhages, multi-organ failure, and death (LINK)…
So, if you’re looking for an LSD-like trip, it’s certainly much safer to stick with… LSD.
I wonder if any effects can be inferred from the parent compound to the nbome. Eg. Are there qualities of 25b-nbome that are reminiscent of 2cb compared to how 2ci correlates to 25I-nbome
I was kind of surprised that 25I-NBMD was as close as they went to FLY-ifying the methoxy-benzyl in the Nichols paper; something tells me that there is potential for some really potent compounds there.