

Designing a Psychedelic State
Intelligent drug combinations to tune subjective experiences.


Peerless psychedelic chemist Alexander Shulgin spent decades tirelessly constructing (and personally assaying) novel psychoactive drugs — by the time of his passing in 2014, aged 88, he had created hundreds of new molecules, many of which have become psychedelic standards and integral components of the pharmacopeia of any psychonaut worth their salt. Although, largely owing to the work of David Nichols’ group at Purdue, we now have a much deeper and more complete understanding of the interactions between drug molecules and endogenous receptors, Shulgin’s “make ’em and taste ’em” approach to drug design remains, in many ways, unbeaten. Whilst rodent assays are fairly effective in predicting which molecules are likely to be broadly psychedelic, it’s only when we get a molecule into the brains of willing volunteers that we really begin to understand the unique constellation of nuanced subjective effects that distinguishes it from other psychedelics. And, even then, explaining why a particular molecule has a particular flavour of psychedelicity remains a largely unsolved problem. If you were to ask a crackajack team of medicinal chemists, pharmacologists, and neuroscientists to design a molecule with effects identical to 5-MeO-DMT (assuming this molecule wasn’t already known), they would likely shake their heads in bewilderment. Where to even start?
The problem is simply stated, but fiendishly difficult to resolve: The overall effects of a particular molecule depend upon the range of different receptors to which it binds and has activity, as well as the manner in which it activates these receptors. But these receptors aren’t distributed equally across the brain: different neuron types, located in different areas of the brain, express different receptor types. Every neuron is itself a complex system, the function and behaviour of which emerges from a vast set of interactions between a multitude of subcellular components and intracellular signalling molecules. Each receptor subtype, when stimulated, perturbs this signalling network in its own unique manner and elicits a specific effect on the neuron. The overall effect of a molecule on a particular neuron will depend upon both the receptor subtypes with which the molecule interacts and the neuron’s particular pattern of receptor expression. Put simply, the effect of a molecule on one neuron type might be quite different to its effect on other neurons, which could be located in completely different areas of the brain. And, of course, all of these different neurons form the bewilderingly complex network from which this unified functional structure, the human brain, emerges. Trying to predict — or engineer — effects on neural activity at the cortical level of molecules that interact with the brain at the subcellular level rarely succeeds beyond tentatively placing the molecule into some broad psychoactive category — “Possibly psychedelic.”

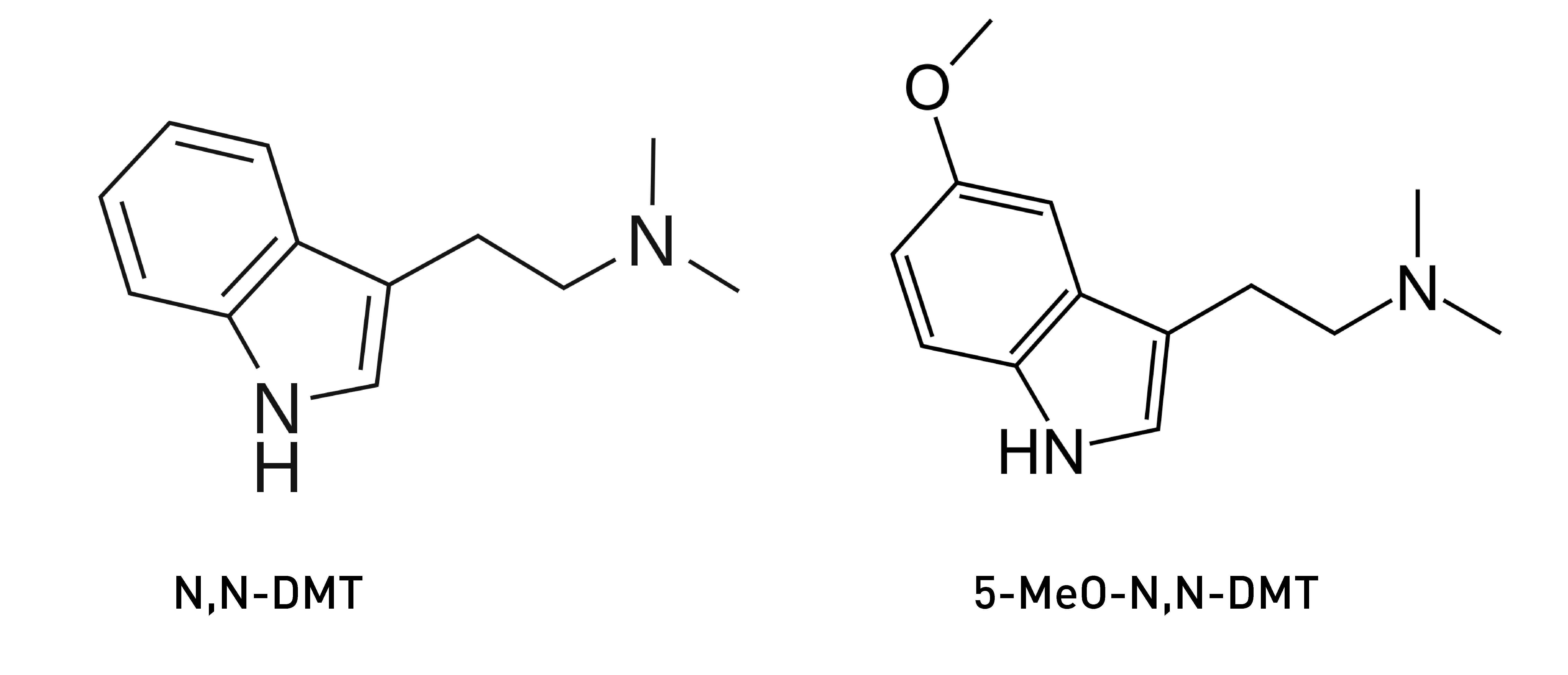
Owing to this layered complexity, it isn’t unusual for closely-related psychedelic molecules to have dramatically different effects. Perhaps the most startling example of this is N,N-DMT vs 5-MeO-DMT. Whereas the former is characteristically extremely visually rich, hurtling you into bizarre inordinately complex worlds filled with intelligent beings of varying form and character, the latter is often described as lacking any striking visual character at all and more akin to a formless void of infinite consciousness and white light.
Although a definitive explanation for these markedly different effects remains out of reach, all is not lost, and whilst there is much we don’t understand, there’s also a lot that we do. But we’re still far from being able to design a molecule with a particular constellation of subjective psychedelic effects. Perhaps this traditional Shulgin-esque model, focusing on the design of single molecules to achieve certain effects, isn’t the optimal approach after all. Let me explain.
Arguably the most important and foundational result to come out of basic psychedelic neuropharmacology research in the last century is that the effects of the classic psychedelics depend primarily upon their activity at the serotonin 5HT2A receptor. When a classic psychedelic binds to this receptor subtype, it increases the excitability of the neuron, making it more likely the neuron will fire and increasing the flow of information between neuronal networks. I’ve discussed in an earlier post how this gives rise to the characteristic changes in the structure and dynamics of the experienced world (and the self). (If you want to get into this much more deeply, then my latest book Reality Switch Technologies is what you’re looking for: LINK).
However, all known classic psychedelic molecules also have varying activity at several of the other 16 or so known serotonin receptor subtypes:
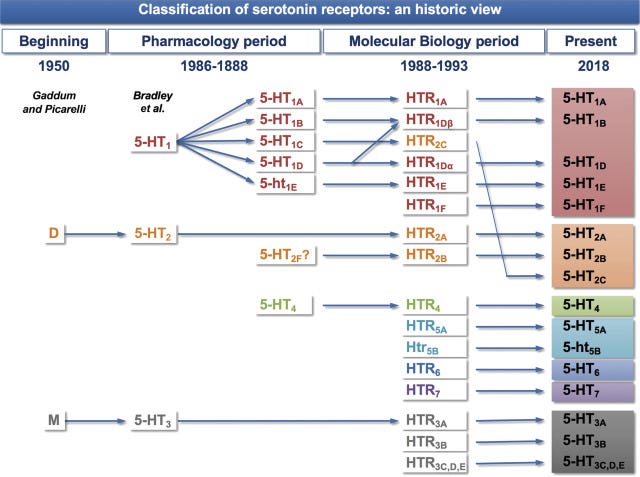
The 5HT1A receptor is particularly interesting, since its effects on neural activity are in direct opposition to those of the 5HT2A receptor. That is, the 5HT1A receptor decreases neuron excitability and opposes the excitatory effects of the 5HT2A receptor. This suggests that, together, these two receptor subtypes form a kind of neural activity “tuning dial”, with the overall excitability of a neuron depending at least partly on the balance of 5HT1A and 5HT2A receptor activation. Many classic psychedelics have some activity at the 5HT1A receptor, which can modulate their effects on neural activity and, as a result, their subjective effects.
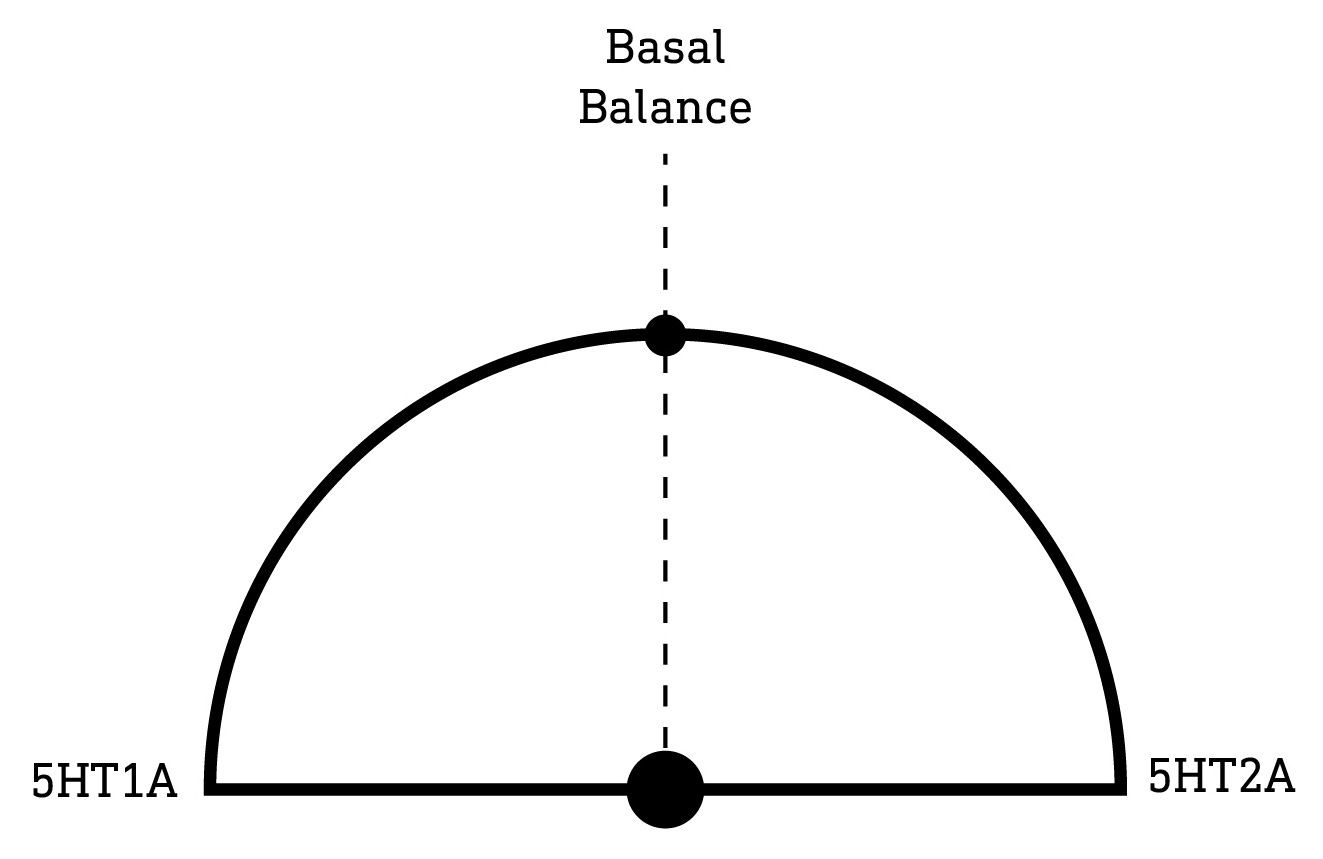
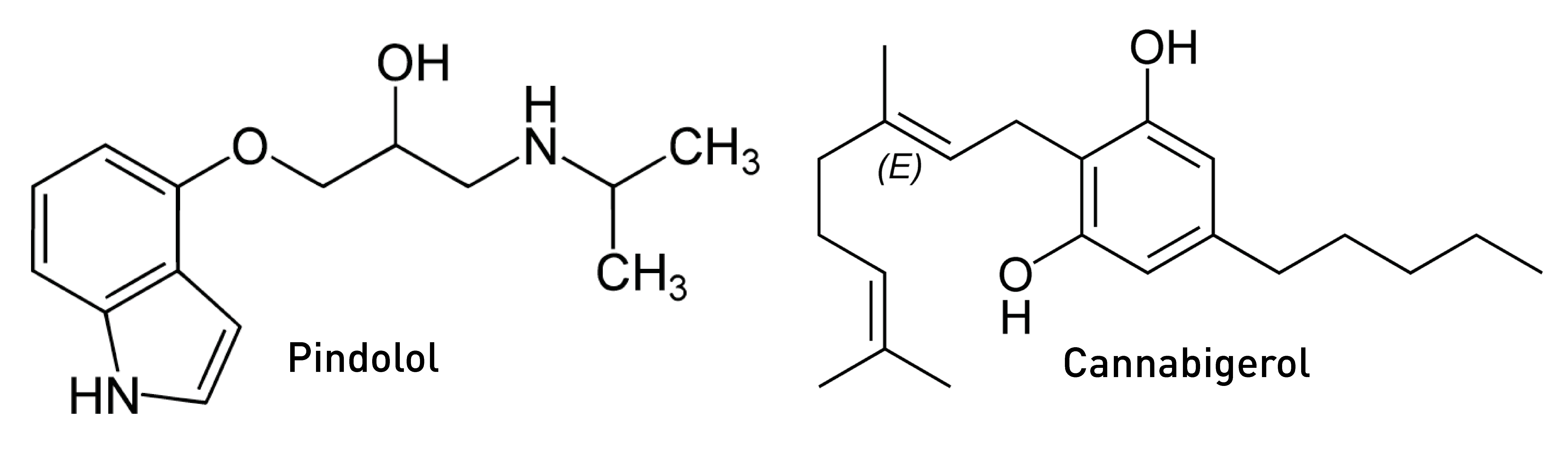
In his pioneering DMT study in the early 1990s, Rick Strassman was well aware of this 5HT1A-5HT2A receptor antagonism and sought to test whether blocking 5HT1A receptors using the 5HT1A antagonist pindolol would enhance the subjective effects of DMT. Strassman surmised that some level of basal 5HT1A activation by endogenous serotonin, as well as DMT itself having some activity at these receptors, likely buffers DMT’s excitatory effects at the 5HT2A receptor. As such, blocking the 5HT1A receptor would eliminate this opposition and, in theory, enhance DMT’s effects. And, indeed, this is precisely what he observed. In fact, Strassman’s subjects described a 2-3 fold increase in the intensity of DMT’s effects when combined with pindolol compared to DMT alone — subjective reports that were borne out by measurements using his Hallucinogen Rating Scale (LINK).

Pindolol is primarily used as a nonselective beta blocker (as an antagonist at adrenergic beta receptors) in the treatment of high blood pressure, with its 5HT1A antagonism being “off target”. Propranolol, another commonly used beta blocker, also antagonises 5HT1A receptors, albeit somewhat more weakly than pindolol. As such, anyone using either medication should tread carefully with DMT — its effects might be much more intense than expected for a given dosage. Interestingly, the natural cannabinoid, cannabigerol, is also a moderate 5HT1A antagonist, but whether this is significant at the levels present in cannabis to modulate the DMT experience is unclear.
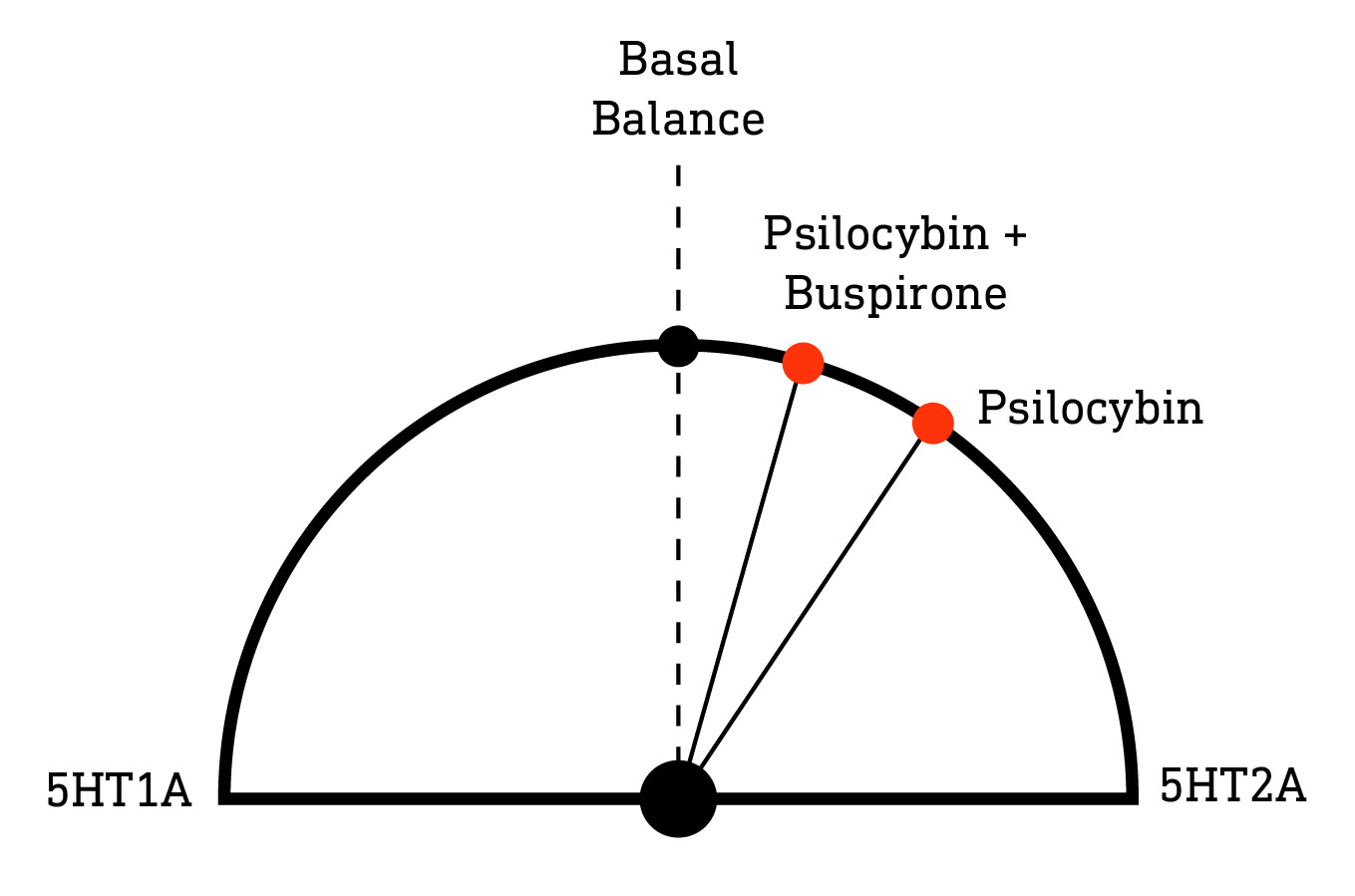
Since 5HT1A antagonists have been shown (at least with DMT) to increase the subjective intensity of psychedelic 5HT2A agonists, we might expect a 5HT1A agonist to nudge the dial in the opposite direction. Buspirone, for example, is a widely-prescribed 5HT1A agonist used to treat anxiety disorders and, by this logic, ought to dampen the subjective effects by enhancing 5HT1A’s opposition to 5HT2A activation. And, in humans at least, this appears to be the case: Franz Vollenweider’s group gave either psilocybin alone or combined with buspirone to almost 20 volunteers and observed a dramatic reduction in the subjective intensity in the latter group (LINK) as measured by the 5D-ASC (Altered States of Consciousness) questionnaire.
The LSD analogue, lysergic acid morpholide (LSM-775), differs structurally from LSD in sporting a morpholine in place of the diethyl group — if you compare LSD and LSM-775, you’ll notice the difference being the bridging of the ethyl groups to form the morpholine ring:
Compared to LSD, LSM-775 has much more potent agonist activity at 5HT1A receptors, in addition to its primary effect at the 5HT2A receptor. In fact, in mice, the only way to elicit the head twitch response (a standard model of psychedelic activity in rodents) is to administer LSM-775 together with a 5HT1A antagonist, thus blocking 5HT1A’s opposition of 5HT2A (LINK).
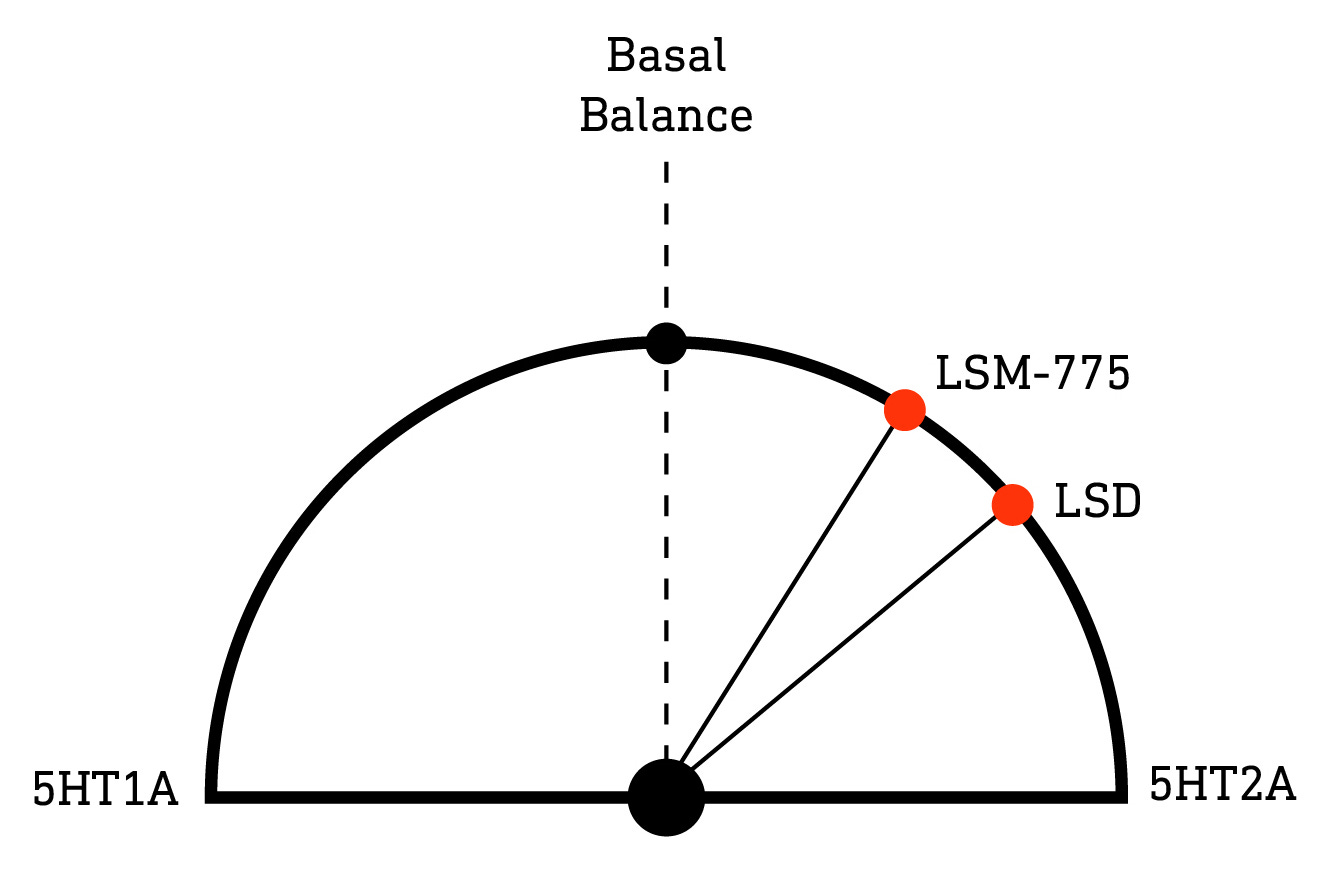
Predictably, based on this animal data, in humans, LSM-775 appears to be much less potent and shorter-acting than LSD. A 1957 study examined its effects in a range of animals — the results being of limited utility — as well as humans (unfortunately only two). The volunteers described 75 micrograms of LSM as being comparable to 50 micrograms of LSD (administered separately beforehand for comparison). However, the effects manifested after around 45 minutes and only lasted for about an hour — obviously a much shorter duration than LSD (LINK).
According to Shulgin:
“There are conflicting reports; one states that 75 micrograms is an effective dose, comparable to a similar dose of LSD, and the other stated that between 350 and 700 micrograms was needed to elicit this response, and that there were fewer signs of cardiovascular stimulation and peripheral toxicity.”
Despite being invented several decades ago, LSM-775 remains a somewhat obscure and rare psychedelic. However, it demonstrates how the level of 5HT1A receptor activation might be adjusted to manipulate the intensity of the psychedelic state. This can be achieved by designing (or discovering) molecules with varying activity at 5HT1A vs 5HT2A receptors (as with LSM-775) or, as with pindolol, employing separate molecules to adjust 5HT1A receptor activation.
However, bearing in mind the complexity I discussed earlier, we’d do well to not get carried away with ourselves here: Although the 5HT1A and 5HT2A receptors oppose each other when expressed in the same neuron, this “tuning dial” is far more than a simple volume control. Compared to regular old N,N-DMT, 5-MeO-DMT has much higher activity at the 5HT1A receptor and, although its psychedelic effects (just as with other classic psychedelics) can be blocked by a 5HT2A antagonist (LINK), the role of 5HT1A receptors in the effects of 5-MeO-DMT appears more complex. Blocking 5HT1A receptors in rats — using the 5HT1A antagonists WAY-100635 or pindolol — at least partially blocked the effects of 5-MeO-DMT (opposite to what Strassman observed with N,N-DMT), although this has never (as far as I’m aware) been tried in humans (LINK). A similar effect has been observed (again, in rats, not humans) with both LSD (LINK) and DPT (N,N-dipropyltryptamine) (LINK).
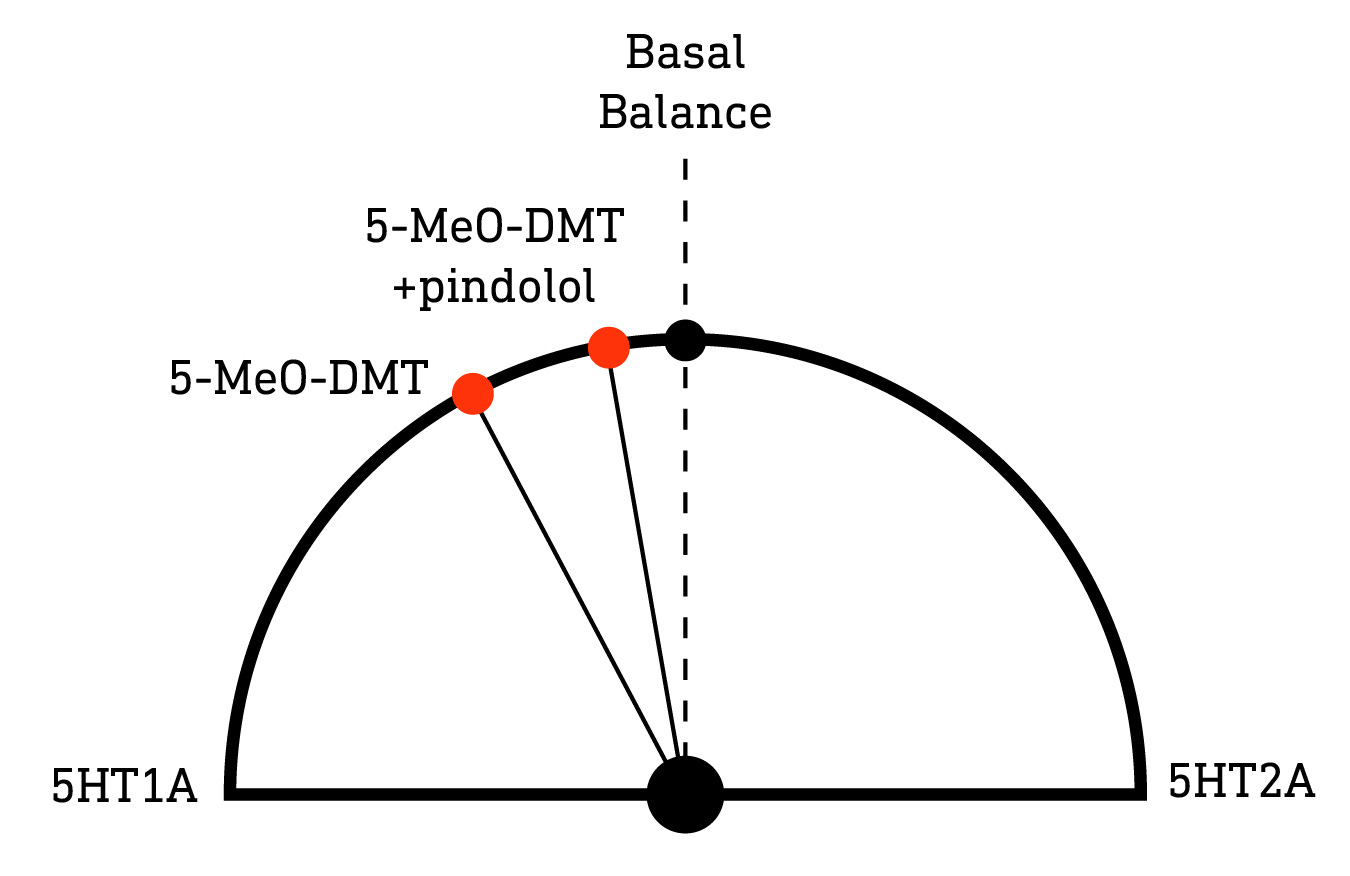
More recent studies suggest that differing expression levels of 5HT1A and 5HT2A receptors in different areas of the brain and in different neuron types are at least partly responsible for this complexity: 5HT1A receptor activation elicits distinct effects on neural activity in different cortical and subcortical regions depending on the local expression of the receptors and, together with 5HT2A activation, the effects of 5-MeO-DMT on overall cortical activity emerge (LINK). In other words, it’s complicated.
So, rather than simply being a volume control, it appears that the balance of 5HT1A vs 5HT2A activation is important not just in regulating the intensity of the subjective effects, but also the type of effects experienced. 5-MeO-DMT achieves its effects by stimulating a highly specific pattern of receptor activity, with 5HT1A activation being no less important than 5HT2A activation. The effects of N,N-DMT, on the other hand, seem to have little dependence on 5HT1A activation and this receptor merely serves to buffer the excitatory effects of the 5HT2A receptor when expressed in the same deep layer pyramidal neurons. So, we might ask, can we “convert” N,N-DMT to 5-MeO-DMT by combining it with a selective 5HT1A agonist? Perhaps the antidepressant alnespirone would do the trick, although we can’t rule out a significant role of other receptor subtypes, so I’m neither suggesting you try this nor that, if you do, it will have the desired effects. I’d still be interested in knowing what happens though.
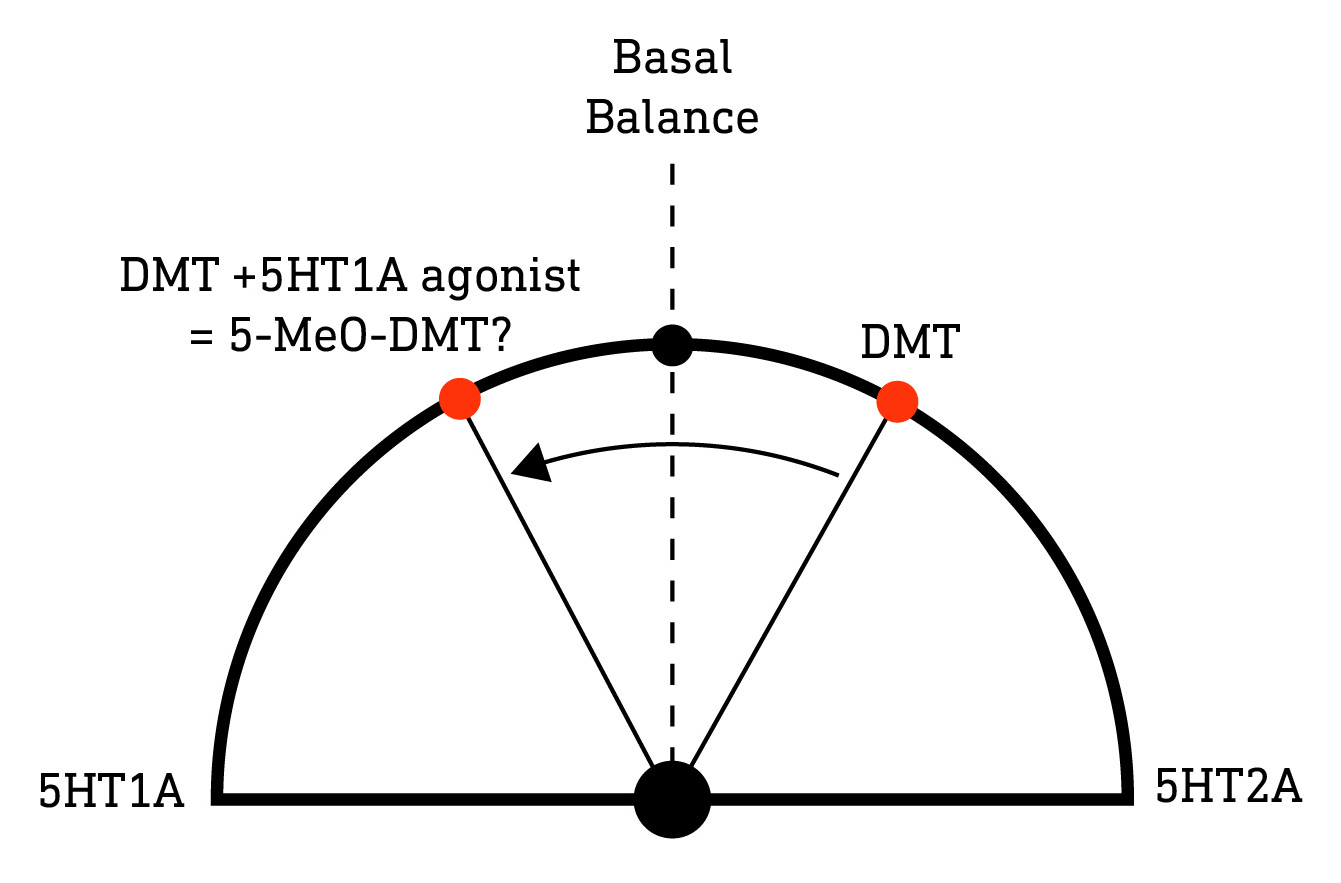
Manipulating receptor activation patterns like this has interesting implications for how we think about drug combinations. When combining drugs with largely independent mechanisms of action and effects — such as LSD with MDMA (“Candy Flipping”) — it’s reasonable to expect subjective effects that feel something like a combination of both drugs (although even this is something of a simplification). However, if you combine drugs with strongly overlapping sites of action and effects, such as a heady hyperspatial mixture of N,N-DMT and N,N-DPT, it would be unwise to assume this to result in a kind of combined effect of both drugs. Rather, what you’re doing here is stimulating a particular balance of 5HT1A and 5HT2A (and other subtype) receptor activation that’s completely different to either drug. Using our tuning dial metaphor, you’re shifting the dial to a position it would never occupy when taking either drug alone. Add a third classic psychedelic to the mix and you’re likely to be in unchartered — and highly unpredictable — territory. This should serve as both an inspiration and a warning.
The key point here is that it isn’t the molecule in itself that elicits a particular subjective effect but, rather, the unique pattern of receptor activation it stimulates. In other words, if two structurally different molecules elicit the same pattern of receptor activation, then we can expect their subjective effects to be the same. Structurally different, but functionally identical. Likewise, it makes no difference whether we use a single molecule with multiple receptor activity to achieve a particular pattern of receptor activation or multiple molecules each with highly selective single receptor activity. Of course this ignores differences in rates of absorption and metabolism, etc, that might affect the experience, so there’d be a lot of work to do if this idea was to be developed practically. However, perhaps the traditional approach to inventing new psychedelic drug effects, by designing single molecules with activity at several receptor subtypes, isn’t the most expedient one. Simultaneously optimising a single molecule with a desired pattern of activity at several receptor subtypes is no easy task and, in many cases, practically impossible to achieve. However, designing a number of molecules, each with highly selective activity at a single receptor is a much more tractable problem. The effect of a single molecule with broad receptor activity could then be reconstituted by several molecules with highly selective activity.
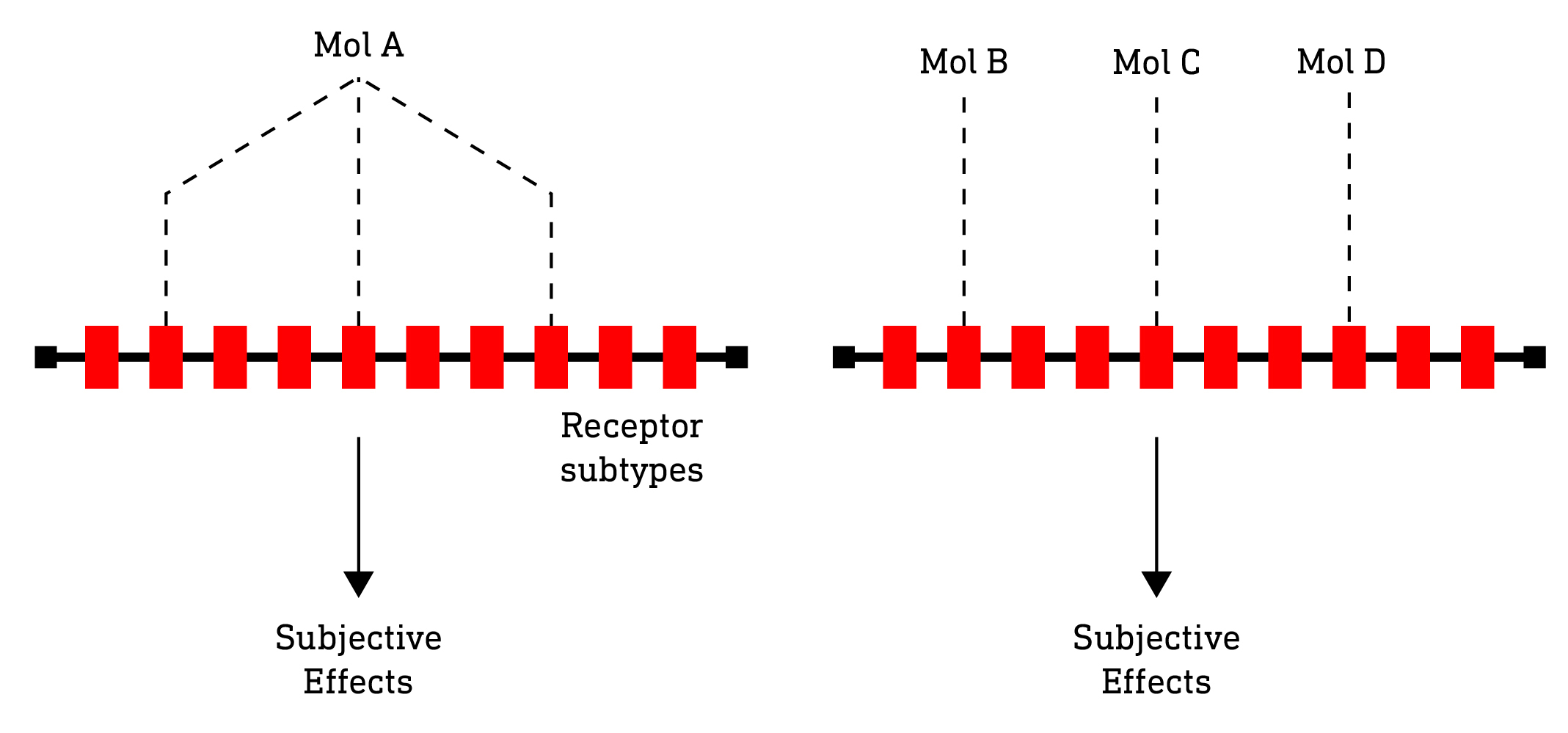
So, all of this naturally leads us to what we might call “Drug Decomposition-Reconstitution”: In much the same way that a particular colour paint can be decomposed into its primary components, a drug can be decomposed into a set of primary receptor interactions. And, as a painter might reconstitute a particular colour from a small set of primary paints, a psychonaut might do likewise with a small palette of highly selective receptor agonists and antagonists, which could be purchased or otherwise procured as a set and, when the effects of a particular drug were desired, it would simply be a matter of looking up the particular combination of primary molecules in a kind of master index. And, of course, you wouldn’t be limited to indexed combinations mapped to known effects: you could just as readily design your own combinations and share your results with friends, family, and fellow psychonauts for them to try. Who knows, you might just stumble upon the next 2C-B…
Great article as always! What would be your thoughts on wether the duration of NN-DMT could be extended by pindalol or some other beta blocker or other molecule? Wondering if the subjective experience would differ from extending the state via MAO inhibition….
beautifully written.. "psychedelicy" 🤌🏼
I have even been thinking about the other less talked about receptors.. for instance.. should we also minimize activation of the 5HT2B receptor in this cocktail? Or should we just add a 5HT2B antagonist? I love the example of 5-MeO vs N,N but I often wonder what is happening with the other 14 receptors? This is a very promising strategy for "removing the trip" but I wonder if the neurobiology is still the same? Would a dampened trip still have the beneficial neuroplasticity?