

Psychedelics, neuroplasticity, and intracellular 5HT2A receptors...
An idiot’s guide to *that* psychedelic neuroplasticity paper...


Whilst my Twitter DMs are usually filled with strangers asking me to interpret their DMT visions (please don’t do this), this week was different. Instead, everyone was asking, “Have you seen this?!?!”, referring to a paper that appeared in the journal Science and was followed by a flurry of news articles breathlessly announcing that a brand new, never-before seen, mechanism of psychedelic action had been discovered. But has it really? Well, it depends what you mean by “psychedelic action”….
Let’s see….

Before we get to that, I thought it would be a good idea to go through the paper in a step-by-step manner, such that those unfamiliar with technical academic neuroscience papers can understand what was discovered and what wasn’t.
Here’s the link to the original paper:
https://www.science.org/doi/10.1126/science.adf0435
First, some skeletal background (this post is long enough already):
The classic psychedelics and ketamine induce neuroplasticity, albeit by different mechanisms.
What is neuroplasticity? In this case, stimulation of the growth of neuronal dendrites.
The classic psychedelics induce neuroplasticity by activation of 5HT2A receptors (the same target of the psychedelic effect).
Although it remains poorly understood, activation of 5HT2A receptors leads to activation of downstream signalling pathways inside the neuron that are ultimately responsible for the growth of dendrites.

The ability of a molecule to induce neuroplasticity requires 5HT2A receptor activation but does not require psychedelic effects. In other words, some molecules induce dendritic growth by activating 5HT2A receptors, but don’t induce psychedelic effects. The authors of this paper surmised that this was because of an effect called location bias: some molecules induce neuroplasticity, not because of some unique kind of activity at 5HT2A receptors, but because they selectively activate receptors in a particular location in the neuron.
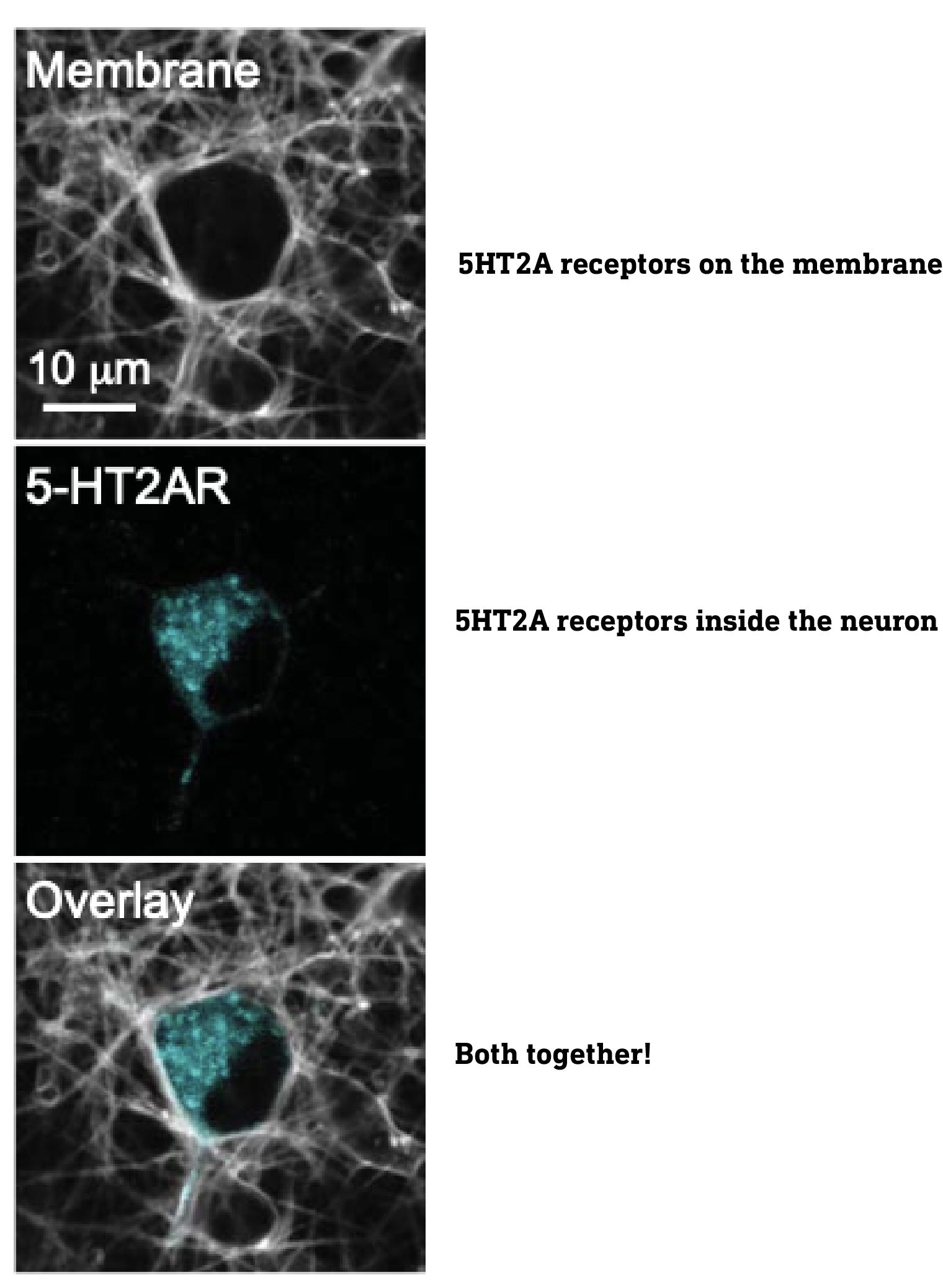
Although most are familiar with receptors embedded on the cell surface plasma membrane, receptors are also found inside the cell, on internal membranes, such as the Golgi apparatus, nuclear membrane, etc. Indeed, when they imaged the 5HT2A receptors using special fluorescent tags, they found large populations both on the surface and deep inside the neurons, particularly in the Golgi apparatus. Of course, to activate these intracellular receptors, a molecule must find its way into the neuron.
This leads directly to the central point of this paper: only the intracellular 5HT2A receptors are responsible for the neuroplasticity effect, so whether or not a molecule that activates 5HT2A receptors induces neuroplasticity depends on whether or not it can reach these intracellular receptors.
The experiments were performed both on isolated neurons in culture (in vitro), as well as in living mice (in vivo). After administering a molecule, they would wait for any neuroplasticity effects to manifest and then measure the increase in density of dendrites in these neurons — this is the measure of a neuroplasticity effect.
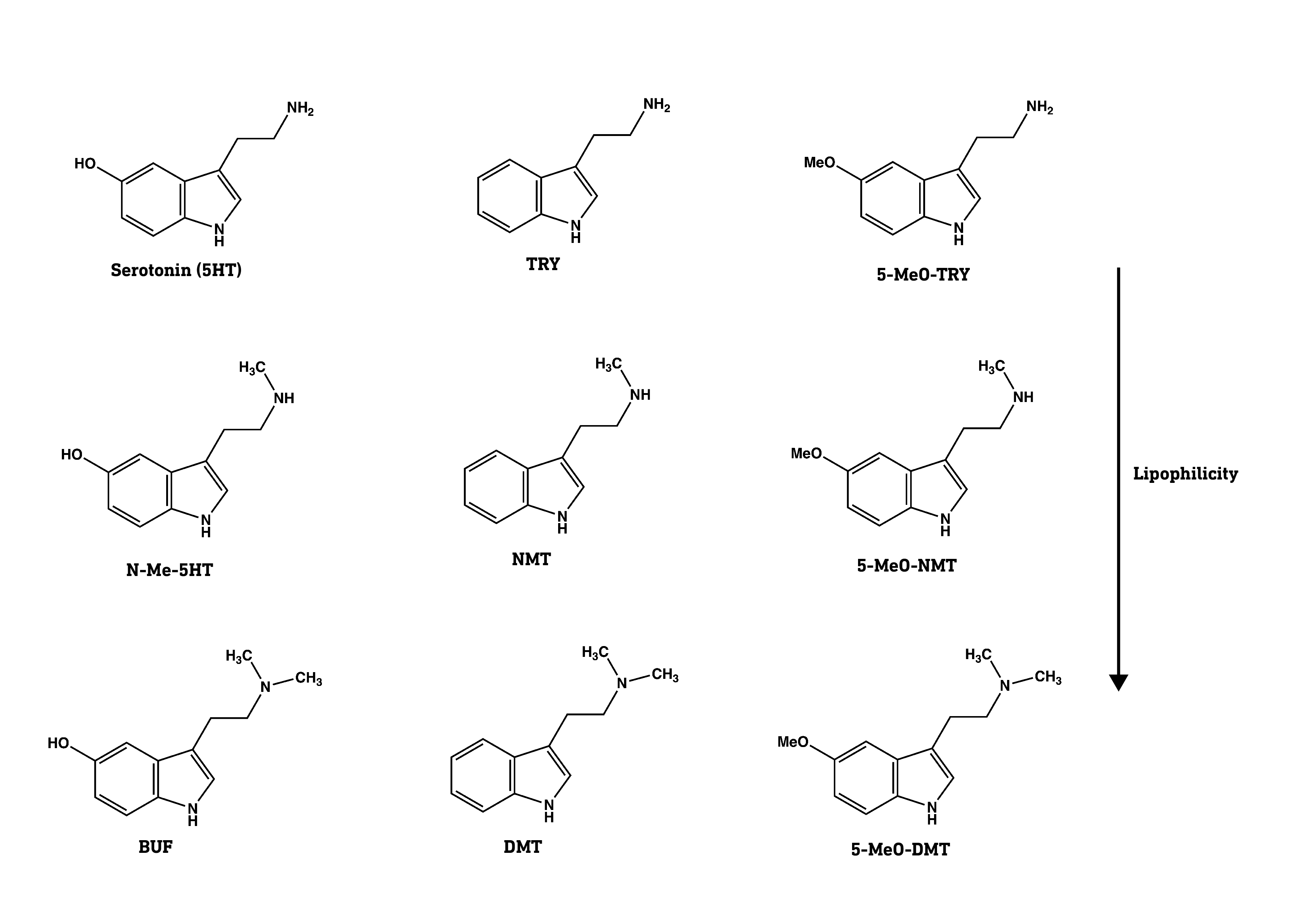
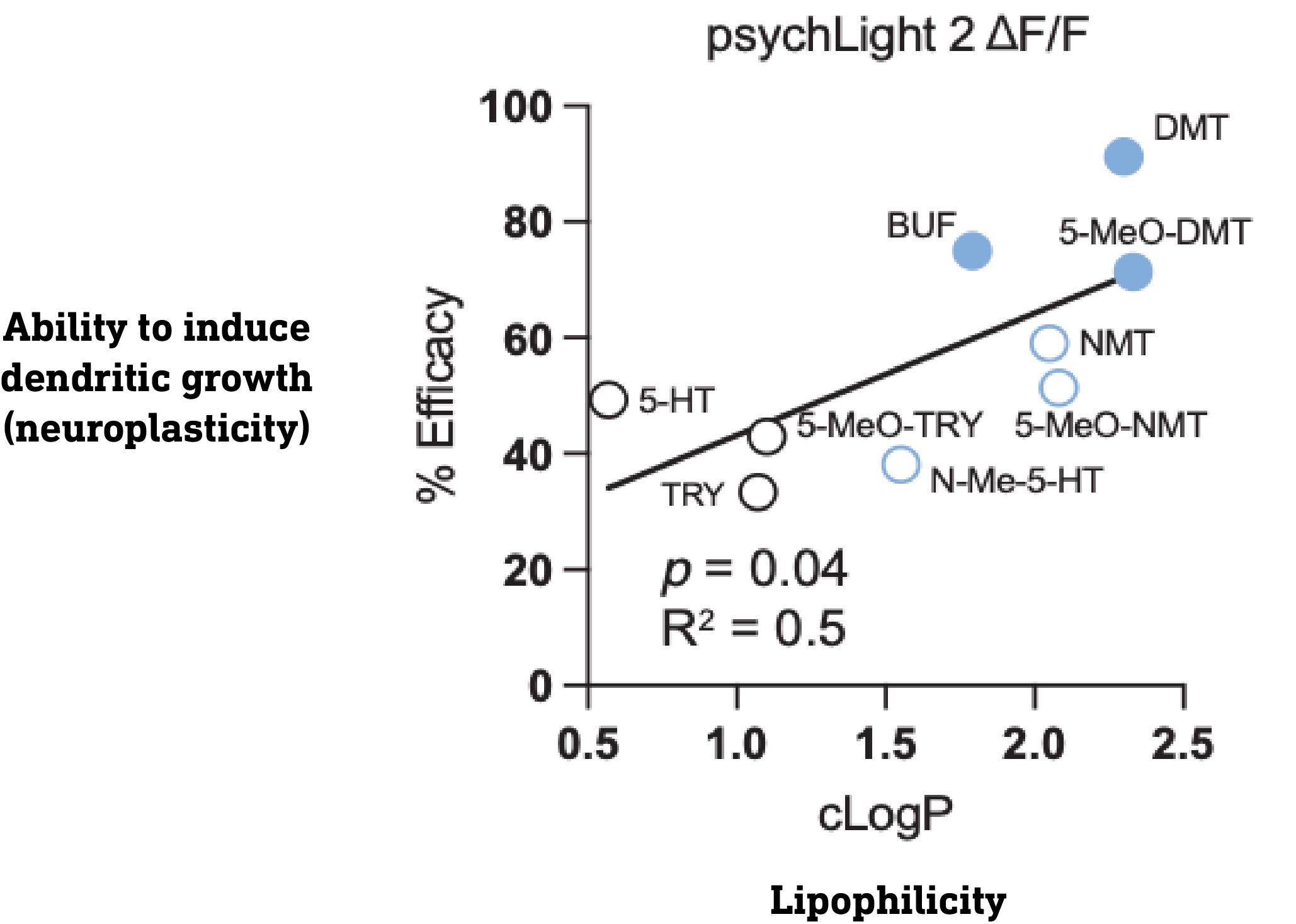
They first tested a series of molecules based on serotonin (5-hydroxytryptamine, 5HT), and the classic psychedelics 5-MeO-DMT, and DMT. Note that both 5-MeO-DMT and DMT have two methyl groups on the side-chain amine nitrogen, whereas serotonin has none. Methyl groups mask the polarity of the amine nitrogen and make the molecule more lipophilic (fat soluble) overall. The authors created a set of molecules based on 5HT, DMT, and 5-MeO-DMT with varying levels of methylation at the amine nitrogen, thus generating molecules with differing levels of lipophilicity. This allowed the authors to look for a relationship between the lipophilicity of a molecule and its ability to promote dendritic growth.
What they found was that the ability to promote neuroplasticity didn’t depend on how well a molecule bound and activated the 5HT2A receptor but, rather, how lipophilic (fat soluble) the molecule was. Why is this significant? In order to enter the neuron, a molecule must pass through the fatty plasma membrane, and only lipophilic molecules are able to do this efficiently — polar (non-lipophilic) molecules cannot. So, this result hints that the molecules are inducing neuroplasticity by acting at 5HT2A receptors inside the neuron rather than on the outside. But it doesn’t prove it. So how to do so?
First, here’s a diagram of a neuron I’ll be using to illustrate the experiments:

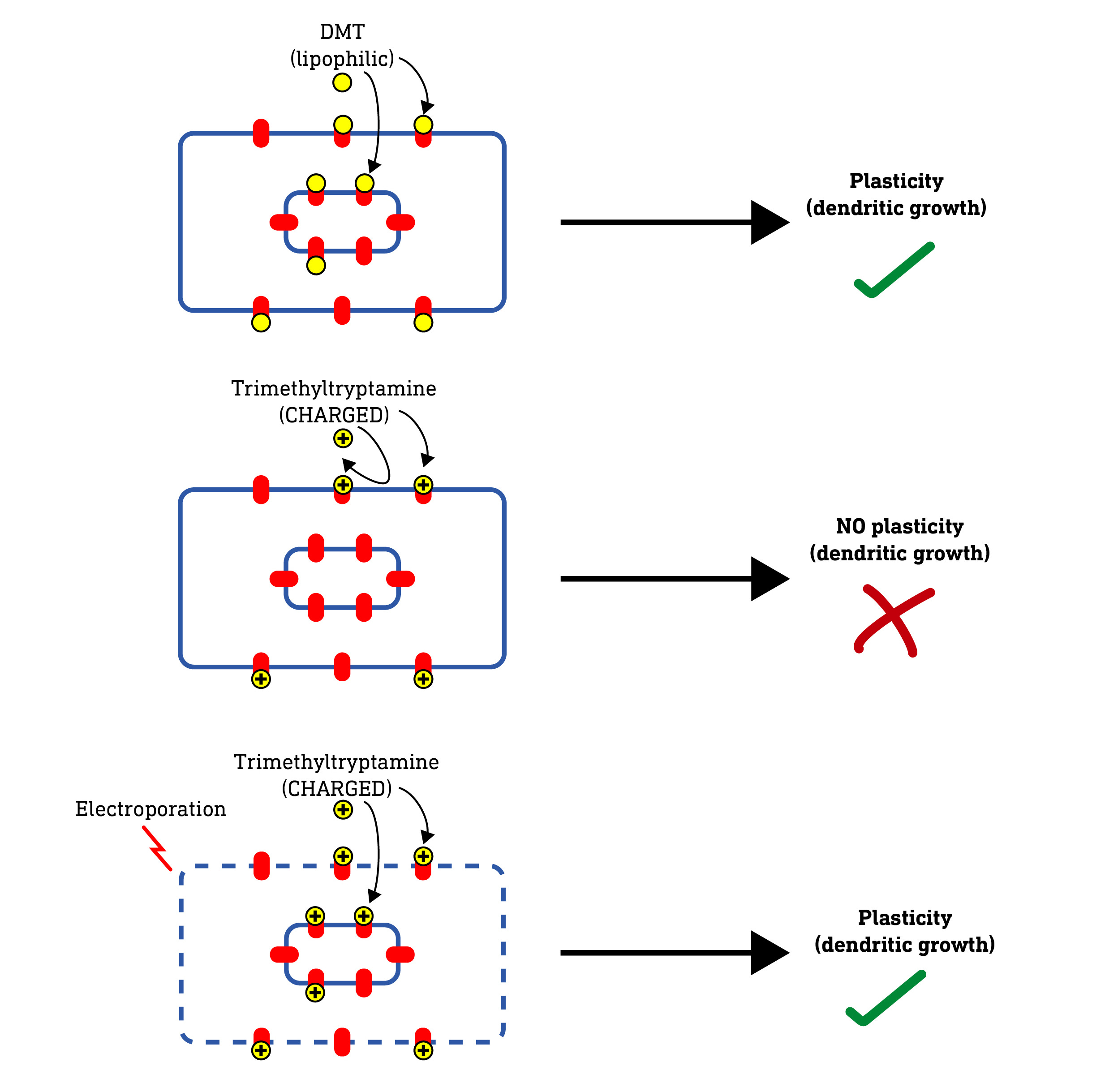
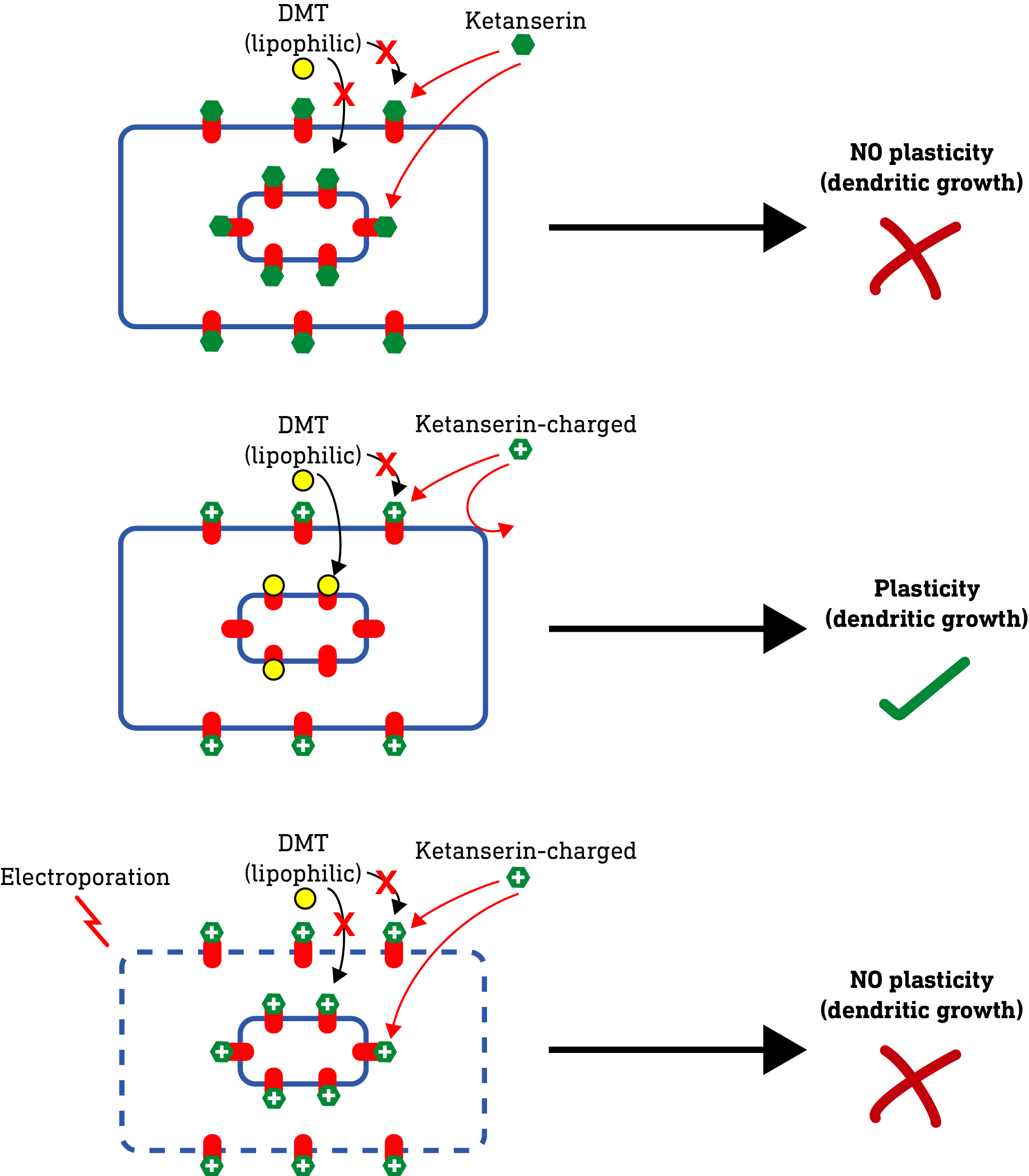
Now, if these intracellular 5HT2A receptors are indeed required for neuroplasticity, a molecule that cannot enter the cell at all — such as a highly polar (water soluble) molecule — should fail to induce dendritic growth. So, they tested this by making a series of molecules with not two but three methyl groups on the amine nitrogen. This forms a quaternary amine that carries a positive charge, making it very water soluble and unable to pass through the cell membrane at all.

When they tested these charged molecules, they found that, although they retained their binding and activity at 5HT2A receptors, they did not induce neuroplasticity. So, it seems the molecule needs to access the intracellular receptors to induce plasticity. However, they weren’t 100% convinced and wanted to be sure that there wasn’t some unexpected effect of changing the molecular structure that blocked neuroplasticity induction.
So, they hypothesised that, if they somehow allowed these charged molecules to enter the neuron, they should regain their ability to induce neuroplasticity. To do this, they used a technique called electroporation, which involves subjecting a neuron to a brief powerful electric charge which temporarily creates microscopic “holes” (pores) in the membrane that allows even charged molecules to pass. As predicted, when applied to neurons subjected to electroporation, the charged molecules were able to induce neuroplasticity — the pores were allowing the molecules to enter the cell and reach the internal receptors. Nice.
Now, the researchers had to be careful here. 5HT2A receptors are not only found inside the neuron but also, of course, on the surface plasma membrane. Whether or not a molecule can get into the neuron has no effect on its binding to these surface 5HT2A receptors. So, how can we be sure that the plasticity doesn’t require activation of both surface and internal 5HT2A receptors?
To test this they used ketanserin, a widely-used selective 5HT2A antagonist (blocker). Crucially, ketanserin is somewhat lipophilic, so it can pass easily through the plasma membrane and, when applied to a neuron, will block both surface and internal 5HT2A receptors. When they applied DMT — a lipophilic molecule that normally induces neuroplasticity — in the presence of ketanserin, it failed to induce dendritic growth, even if the neurons were subjected to electroporation. Obviously, ketanserin was blocking all 5HT2A receptors — both surface and internal receptors — so DMT had no effect. This doesn’t tell us much. However, what if we could block only the surface 5HT2A receptors, whilst leaving the intracellular receptors untouched? If DMT does indeed require only the intracellular 5HT2A receptors to induce neuroplasticity, then it shouldn’t be affected by selective blocking of the surface 5HT2A receptors.
To achieve this, the authors created a charged methylated ketanserin analogue that’s unable to pass through the plasma membrane and, as such, can only block receptors on the cell surface. In the presence of this analogue, DMT was still able to pass through the membrane and induce neuroplasticity by activating the intracellular receptors, demonstrating that only these 5HT2A receptors are necessary for the neuroplasticity effect. However, when the neurons were also subjected to electroporation, the charged ketanserin analogue was able to get into the cell and block the intracellular 5HT2A receptors as well. This, as predicted, prevented DMT from inducing neuroplasticity. So, we can now safely conclude that activation of intracellular 5HT2A receptors alone is responsible for the neuroplasticity effect.
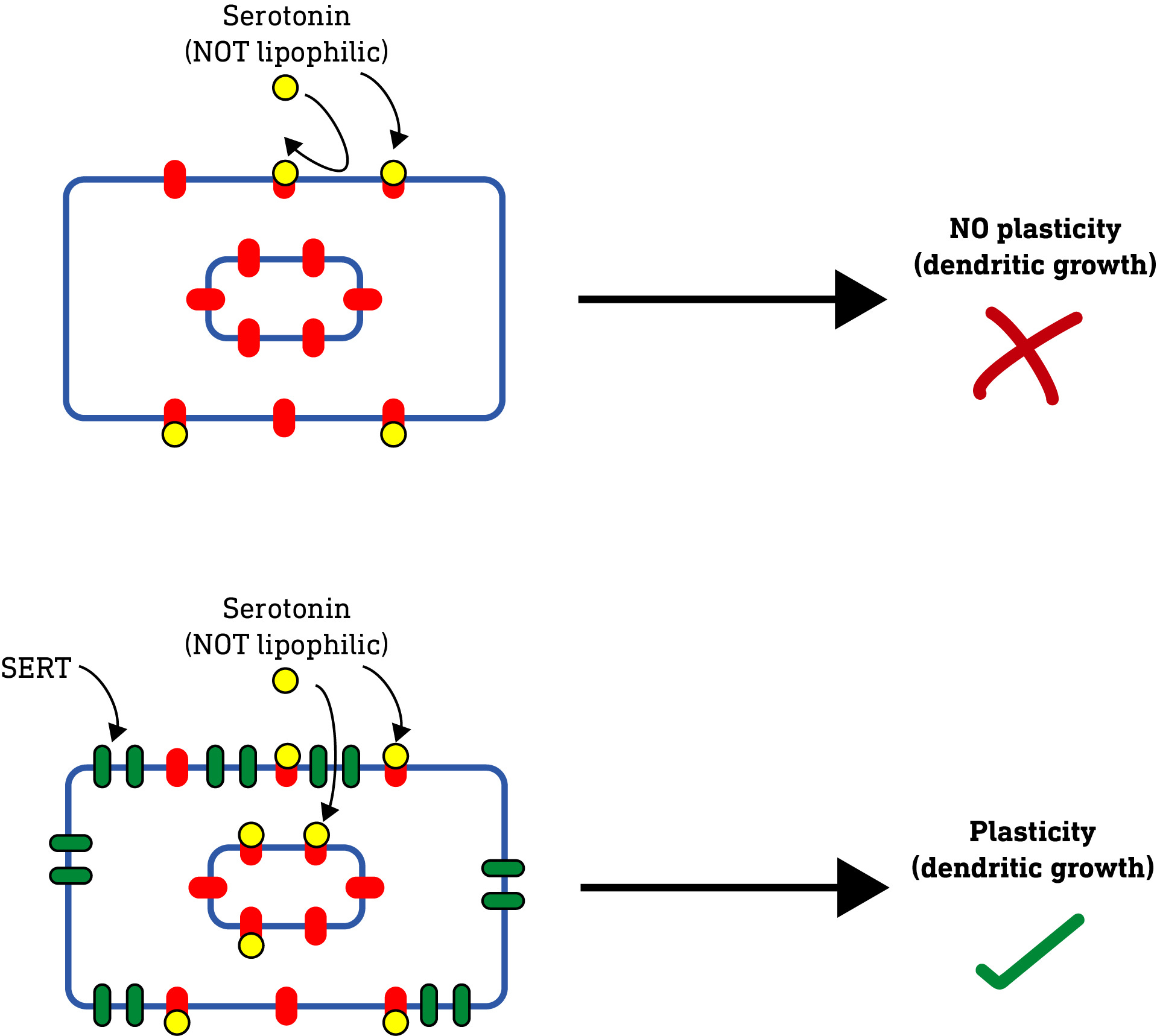
All of these results so far raised an interesting question: Serotonin (5-hydroxytryptamine, 5HT), unlike the classic psychedelics, doesn’t induce neuroplasticity (nor is it a psychedelic). It was always assumed that this was because of how it interacted with and activated the 5HT2A receptor. However, the authors’ results so far suggested that perhaps it was nothing to do with this, but simply that serotonin, being water-soluble and unable to pass through the plasma membrane, simply couldn’t reach the intracellular 5HT2A receptors required to induce neuroplasticity. So, they hypothesised that, if they applied serotonin to neurons subjected to electroporation, thus allowing it to enter the neuron, then serotonin should induce neuroplasticity. And, indeed, this is what they found.
They further confirmed this result by using an alternative to electroporation. Rather than using electricity to make holes in the membrane, they expressed a serotonin transporter protein (SERT) on the surface of the neurons. These SERT proteins are specialised to pump serotonin from the outside of the neuron to the inside. When serotonin was applied to these neurons, just as with electroporation, it induced dendrite growth.
Note that both these experiments with serotonin were performed with isolated neurons (in vitro), so the authors wanted to check that extracellular serotonin in a living brain (in vivo) could also induce dendritic growth if able to get inside of the neurons. To do this, they selectively introduced the SERT transporter (using special techniques I won’t discuss here) into neurons of the prefrontal cortex of mice. They then used a selective serotonin-releasing drug, PCA (para-chloroamphetamine), to raise the levels of extracellular serotonin in the mouse’s brain, predicting that the serotonin would be transported into the neurons via SERT and induce dendritic growth. And, indeed, this is what they found. Furthermore, the mice also demonstrated an improvement in the “forced swim test”, a standard procedure used to test for anti-depressant activity in novel molecules.
So, overall, we now have multiple lines of evidence showing that a 5HT2A-activating molecule will induce plasticity, whether or not it is psychedelic, as long as it can reach and activate the intracellular population of 5HT2A receptors, in both isolated neurons and living animals. Furthermore, the forced swim test result suggests that this dendritic growth could map to improvement in symptoms of depression. Major results complete.
Now we get to the murkier and more controversial result in the paper.
We know that serotonin binds and activates 5HT2A receptors, but doesn’t normally cause a psychedelic effect, whereas a molecule like DMT or psilocin most certainly does. This is thought to be because of functional selectivity, which refers to the manner in which a molecule activates the receptor and elicits downstream signalling inside the neuron. Only psychedelic molecules are able to activate the 5HT2A receptor in such a way that psychedelic effects manifest. Why this is the case is complicated, but has to do with the distinct pattern of intracellular signalling elicited by psychedelic 5HT2A agonists (such as DMT) compared to their non-psychedelic counterparts (such as serotonin).
The standard test for psychedelic effects in mice is the head twitch response (HTR): psychedelic 5HT2A agonists (such as psilocin) induce a characteristic twitching movement of the mouse’s head, but non-psychedelic agonists (such as lisuride or serotonin) do not.
When the authors increased brain serotonin levels using PCA in normal mice, there was no HTR response. No surprises there. However, in mice fitted with the SERT transporter (that pumps serotonin into neurons), increasing serotonin, unexpectedly, induced a HTR, suggesting it was eliciting psychedelic effects. The author are suitably circumspect in drawing conclusions here:
“Activation of intracellular cortical 5-HT2ARs may play a role in the subjective effects of psychedelics.”
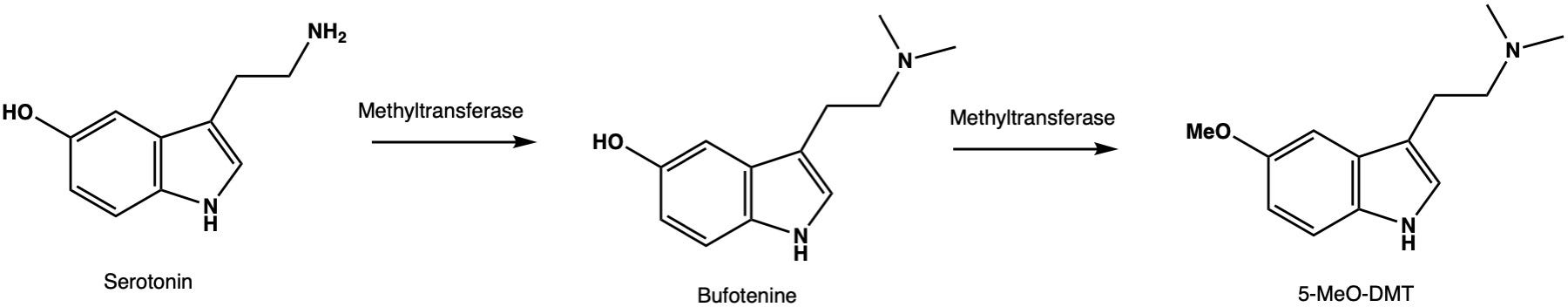
This result isn’t entirely new: It has previously been shown (in a paper published in 2010 (LINK) that, if you ramp up the levels of serotonin in a normal mouse brain sufficiently, you will get a HTR (i.e. an indicator of psychedelic effects), even if the serotonin isn’t provided with a passage into the neurons. Why? Well, serotonin can be converted to psychedelic N-methylated tryptamines (such as bufotenine and 5-MeO-DMT) by enzymes in the mouse brain. And, when serotonin levels are artificially increased, the levels of these psychedelic tryptamines also increases to a level sufficient to elicit psychedelic effects. It’s these psychedelic methylated tryptamine metabolites that induces the HTR, not serotonin. Indeed, you can block the HTR by inhibiting the methyltransferase enzymes that perform the conversion from serotonin to the psychedelic methylated tryptamines.
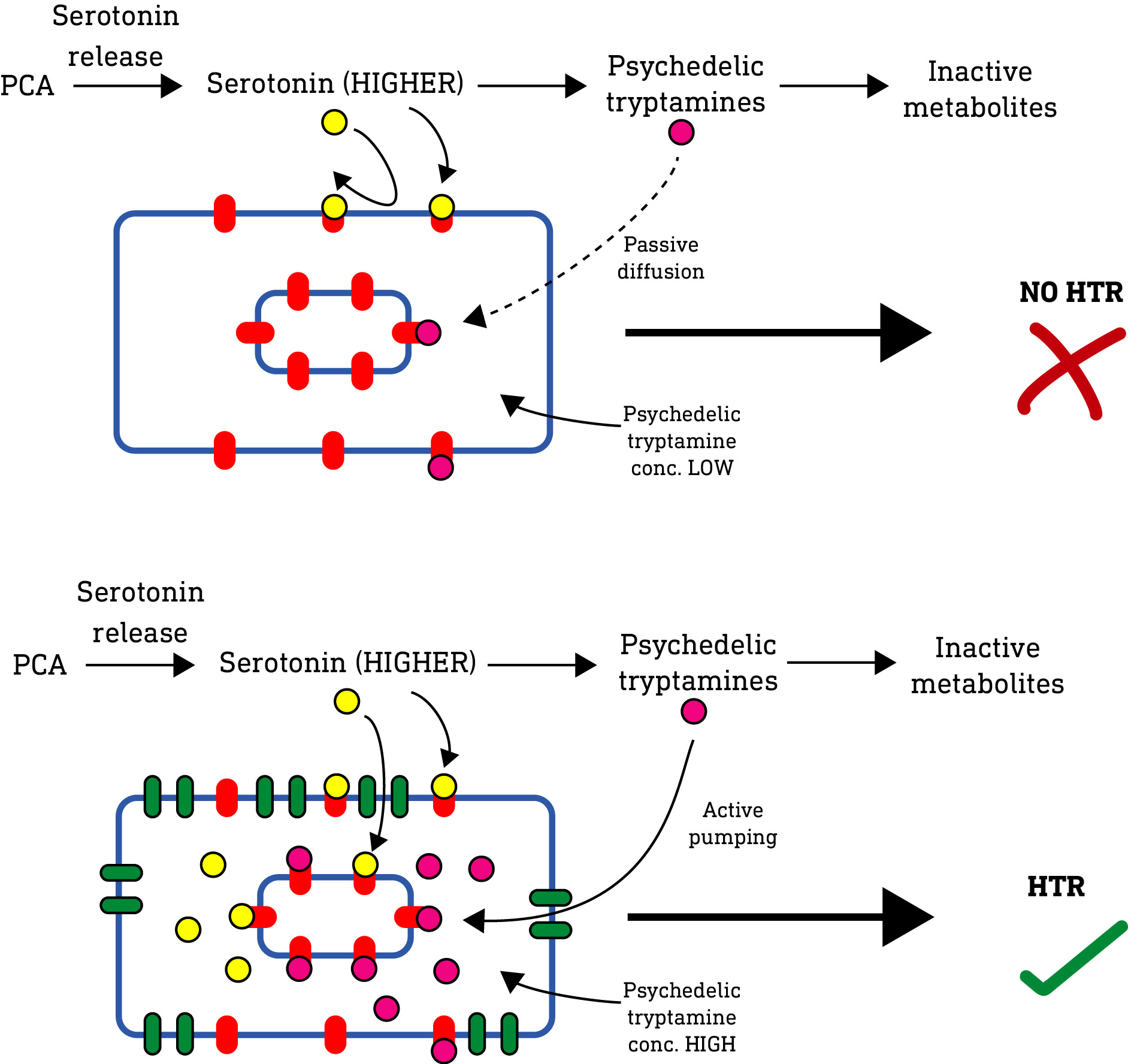
So, what’s going on in this paper? Well, again, they used a drug (PCA) to artificially increase serotonin levels, and so also increased the level of psychedelic tryptamines in the mouse brain. So, just like the 2010 paper, we should expect a HTR. But, something is amiss. Why didn’t the normal mice (i.e. without the SERT transporter) also exhibit a HTR? In the 2010 paper, all that was required to elicit a HTR was to increase serotonin — there was no need to give serotonin access to the intracellular 5HT2A receptors as well as the surface receptors. But in this paper, the SERT transporter was required. What’s going on? Well, my guess is as follows:
In normal mice, PCA simply didn’t push the level of serotonin high enough to generate sufficient levels of psychedelic tryptamine metabolites to elicit a psychedelic effect. So they were quickly metabolised or diffused away without reaching a concentration high enough to elicit a psychedelic (HTR) effect.
Now, the active psychedelic tryptamine metabolites have varying lipophilicity depending on their methylation level, and so varying abilities to pass through the neuronal membrane and accumulate inside the neuron. However, a 2009 paper (LINK) previously showed that psychedelic N-methylated tryptamines are substrates for the SERT transporter, which would allow them to accumulate inside the neuron independent of their lipophilicity. So, in the mice equipped with SERT, even though the levels of psychedelic tryptamine metabolites generated outside the cell was relatively low (and insufficient to elicit a HTR), the SERT transporter actively pumped and accumulated these metabolites inside the neuron, such that they were able to reach a sufficient concentration at the intracellular 5HT2A receptors to elicit the HTR response as observed.
The authors also point out:
“A substantial proportion of 5-HT2ARs in cortical neurons are localized to the Golgi, and intracellular compartments such as the Golgi are slightly acidic compared with the cytosol and extracellular space. Thus, it is possible that protonation of psychedelics within the Golgi leads to retention and sustained signaling, which results in neuronal growth, even after transient stimulation.”
In other words, once a psychedelic gets inside the neuron and the Golgi, it might become “trapped” and accumulate there, allowing it to keep activating the 5HT2A receptor for longer. This could further aid in the accumulation of the active tryptamine metabolites inside the neuron.
So, whilst psychedelics do need to activate intracellular 5HT2A receptors to have neuroplasticity effects, they don’t need to get inside the neuron to have psychedelic effects — they just need to be present (or accumulate) in sufficient concentrations around 5HT2A receptors, whether these are on the membrane or inside the neuron. Equipping mouse brains with SERT simply allowed them to actively accumulate the active psychedelic tryptamine metabolites inside their neurons, despite their low levels of production from serotonin, where they could activate the intracellular 5HT2A receptors at sufficient levels to induce a HTR. In other words, it’s not the location of the 5HT2A receptors that matters for psychedelic activity, just that they’re sufficiently activated by psychedelic 5HT2A agonists. My guess is this HTR result would disappear with an N-methyltransferase inhibitor that blocked formation of psychedelic methylated tryptamine metabolites (as was seen in the 2010 paper).
So, has a new mechanism of psychedelic drug action been discovered? Well, the mechanism of neuroplasticity has been further elucidated. But, I don’t believe that a new mechanism for the psychedelic effects has been discovered. But I could be wrong (and often am).
Amazing breakdown, thank you!
I wonder, why do we have 5ht2a receptors inside our neurons if our natural neurotransmitters like serotonin would never reach them due to their polarity? And why would triggering them cause psychedelic effects / dendritic growth? And are the subjective effects and dendritic growth unrelated effects of the stimulus, or do the subjective effects cause the dendritic growth (or vice versa)?
I'm assuming that when we're young and our brains are developing we experience a lot of dendritic growth- what causes that? Is it activation of the same receptors, and if so what is activating them?
Thank you for doing this!